A phase II study of thalidomide and temozolomide in patients with brain metastases from malignant melanoma: lymphopenia correlates with response
Abstract
Background: Central nervous system (CNS) metastases develop in nearly half of patients with advanced melanoma and in 15–20% CNS is the first site of relapse. Median overall survival is short, ranging from two to four months, and one-year survival rate is only 10–15%. THA has been shown to have both anti-angiogenetic and immuno-modulating effects. TMZ is an oral alkylating agent with an excellent oral bioavailability and it is highly lipophillic with an ability to penetrate the blood–brain barrier. TMZ and THA in combination were tested in patients with brain metastases from malignant melanoma.
Methods: Between June 2004 and February
2007 patients with measurable metastatic melanoma in progression and PS ≤
1 received TMZ in a dose of 150 mg/m2 qd for seven days, followed by
seven days off therapy and THA in 200 mg qd, both orally administered.
Concomitant treatment with steroids was allowed. PBMCs were collected from the
last 14 consecutive patients for evaluation of immune parameters.
Results: Forty screened patients were eligible and evaluable for
response, and 39 were evaluable for toxicity. 25 patients had asymptomatic and
15 symptomatic brain metastases. The toxicity was primarily grade 1–2 with no
grade 4 or treatment-related deaths. Four patients had thromboembolic events
grade 3. One patient obtained a CR and five a PR in the CNS, while two had CR
and four had PR outside CNS. Overall response rate was 17.5%. We found a
significant positive correlation between lymphopenia and objective response.
Conclusions: The combination treatment was well tolerated but with more
frequent thromboembolic events compared to single drug TMZ or THA. The
treatment demonstrated activity in CNS as well as outside CNS. The correlation
between lymphopenia and objective response needs further investigation.
Introduction
Malignant melanoma (MM) in the advanced stage has a poor prognosis. The disease is notorious for its propensity to metastasize and for its poor response to current medical therapeutic regimens, with median survival times in most studies ranging from six to nine months and five-year survival rate of less than 5% [1]. Furthermore, melanoma is the third most common cause of metastases in the central nervous system (CNS) after carcinomas of the lung and breast and develops in nearly 50% of patients with metastatic melanoma. CNS metastases is the first failure site in approximately 40% among the 8–10% of patients with an initial complete response to high-dose IL-2-based therapy [2]. Median survival time in these patients is short, ranging from two to four months, and one-year survival rate is 10–15% [3].
Once patients develop brain metastases, treatment is palliative. Surgery and
stereotactic radiotherapy can produce effective palliation in patients with a
few CNS lesions [4]. In case of multiple or inoperable CNS
metastases, whole brain radiotherapy is considered standard of care.
Temozolomide (TMZ) is an oral, alkylating agent with a high oral
bioavailability. It is highly lipophilic with an ability to penetrate the
blood–brain barrier, and it has demonstrated activity in the treatment of brain
metastases from MM [5]. The schedule for treatment with TMZ
is of utmost importance. Compared to the standard dose of 150–200 mg/m2
five days every 28 days, extended dose permits a 2.1-fold greater drug exposure
over four weeks. Different schedules have been tried [6,7]. A lower dose per day but given continuously over a period
of 6–8 weeks followed by a two-week rest has led to significant toxicity, with
lymphopenia in up to 75% of the patients treated [8,9]. An increased depletion of the DNA repair enzyme
0(6)-alkylguanin-DNA alkyltransferase (AGAT) has been observed with the
extended dosing, which may lead to enhanced anti-tumour activity. In patients treated
with TMZ, the activity of the enzyme was measured in mononuclear cells in the
blood [7].
Thalidomide (THA) was introduced in Europe and Canada in the late 1950s as a
sedative and for the treatment of morning sickness in pregnant women. The drug
was withdrawn from the market in the early 1960s because reports of
teratogenecity associated with its use emerged [10]. D'Amato
et al were the first to show the inhibitory activity of THA on
angiogenesis in a rabbit model of corneal neovascularization induced by basic
fibroblast growth factor (bFGF) and/or vascular endothelial growth factor
(VEGF) [11]. Furthermore, THA inhibits the inflammatory
response by decreasing cyclo-oxygenase-2 activity and by suppressing the
synthesis of tumour necrosis factor-a (TNF-a) [12] by
inducing TNF-a mRNA degradation [13]. THA enhances the
production of IL-2 and IL-2 receptors, modulating the immune system to induce
anti-cancer activity. The function of other cytokines, such as IL-6 and IL-12,
are inhibited by THA, and it also modulates the expression of cell adhesion
molecules [14–16]. THA has
demonstrated clinical activity against a range of solid tumours, including
melanoma [17]. We found in a phase-II trial that THA as
single agent has some extra-cranial activity in patients with brain metastases
from MM but no objective response was seen in the brain [18].
Hwu et al reasoned that additional benefit might be gained by combining
TMZ with THA and that the combination of continuous daily oral TMZ and THA
appears to be a promising oral regimen for the treatment of MM, particularly in
patients with brain metastases [19].
With the intention of optimizing this regimen we have conducted a phase-II
trial with TMZ using extended dose, 150 mg/m2 qd for seven days,
followed by seven days off therapy and THA 200 mg qd.
Materials and methods
Patients
All patients included in the study had brain metastases in progression not amenable for surgery or stereotactic radiotherapy and due to limited symptoms whole brain radiotherapy was not indicated. WHO performance status (PS) ≤ 0.1. Further inclusion criteria were: measurable disease in the brain, defined as ≥ 1.0 cm in diameter on spiral CT or MRI. Concomitant steroids were allowed for patients with clinical evidence of brain metastases. Lesions within a previous radiation field had to have shown recent disease progression to be considered evaluable. Previous treatment with IL-2, other biologic therapy or radiotherapy must have been completed ≥ 4 weeks prior to study enrolment. Eligible patients also met the following criteria: WBC ≥ 3.0 x 109/l, ANC ≥ 1.5 x 109/l, platelets ≥ 100 x 109/l, serum alanine aminotransferase and alkaline phosphatase levels < 2.5 UNL and bilirubin and creatinine ≤ 1.5 UNL. All patients were obliged to comply with Pharmion's Risk Management Programme, conducted according to the System for Thalidomide Education and Prescribing Safety (STEPS) guidelines [20]. Female patients were required to have negative urine pregnancy test, within 24 hours prior to THA therapy. Any women of child-bearing potential had to use adequate contraceptive methods during the study and for 28 days after the last THA dose. Men were required to use condoms in connection with any sexual activity. Pregnant or nursing women were excluded. All patients provided written informed consent.
Treatment and patient evaluation
Starting dose of THA was 100 mg qd orally, taken in the evening to reduce drug-related daytime somnolence. The dose was escalated after one week to a maximum of 200 mg qd given continuously. Concomitantly, patients received treatment with TMZ 150 mg/m2 qd for seven days, followed by seven days off therapy. Treatment was continued until unacceptable toxicity, patient refusal or disease progression.
Toxicity was assessed according to Common Toxicity Criteria (CTC v3.0) [21]. The dose of THA was interrupted in cases of myelotoxicity, constipation or somnolence grade 3–4 and could be resumed if toxicity was reduced to grade 1. In cases of treatment interruption due to THA related myelotoxicity or constipation, somnolence (≥ grade 2) or neuropathy (> grade 2), treatment could be restarted with 50% dose reduction. No dose re-escalation was performed. In cases of grade 4, neuropathy treatment was stopped.
Dose modification guidelines for TMZ allowed for interruption of treatment in the event of any grade 3–4 toxicity, until toxicity resolved to ≤ grade 1 or baseline level. Subsequently, TMZ would be restarted at a dose of 112.5 mg/m2/d. With subsequent toxicity > grade 2 doses would be reduced to 75 mg/m2/d, which was the lowest allowed dose. Re-escalation of the dose was not allowed.
One treatment cycle was defined as a four-week period. Clinical evaluation was performed after the initial two and four weeks in order to perform proper dose correction. Thereafter, patient clinical evaluation was performed at four-week intervals to assess toxicity and clinical benefit. During each visit, a complete blood count was taken, combined with evaluation of blood pressure, weight and performance status.
Response to treatment was assessed after three cycles according to RECIST [22], by physical examination and CT or MRI of all metastatic sites. In case of disease progression prior to first evaluation, treatment was stopped and the patient was designated as having progressive disease (PD). In case of stable disease (SD) or an objective response, partial response (PR) or complete response (CR), treatment was continued.
Identification of lymphopenia
The absolute lymphocyte count (ALC) was determined together with the complete blood count during each visit. Lymphopenia was defined as ALC < 0,8 x 109/l corresponding to CTC grade 2. Severe lymphopenia was considered to be ALC < 0.5 x 109/l (≥ grade 3 lymphopenia). Initially, prophylactic antibiotics were not administered for lymphopenia, but following cases of opportunistic infections occurring among the first 15 patients enrolled in the study. We started to administer prophylactic antibiotics (Sulfamethoxazol/Trimetoprim two tablets three times a week) to patients with grade 2 lymphopenia and fever > 38º and to patients with ≥ grade 3 lymphopenia with or without fever.
Analysis of T-cell populations
Three peripheral blood samples were obtained from the last 12 patients enrolled in the study. One before treatment with THA and TMZ (pre-sample), one after treatment day 14 and one on day 28. Mononuclear cells (MNC) were purified using leucosep® tubes (Greiner Bio-One GmbH, Germany). Red blood cells were lysed and MNCs were cryopreserved in foetal bovine serum (FBS) (Gibco, Invitrogen, Carlsbad, CA, USA) containing 5% DMSO until use. For staining of CD4+CD25+FOXP3+ Treg cells the PE anti-human FOXP3 staining kit from eBioscience was used (eBioscience, San Diego, CA, USA). In brief, cells were stained with FITC anti-human CD127, PE anti-human FOXP3, PerCP anti-human CD4, APC anti-human CD25 and the relevant isotype controls as recommended by the manufacturer. The cells analysed on a FACSdiva (Becton Dickinson). The collection was stopped after acquiring 200,000 events, and data were analysed using FACSdiva software (Becton Dickinson). For defining the population of Treg cells, the cells were first gated on CD4+ lymphocytes then on CD4+ and CD25high. On the pre-sample, the level of CD4+CD25high out of CD4+ T cells was fixed at 2%, and the following two samples (day 14 and day 28) were analysed on the same day using the same gates. To confirm that the cells within the gate were Treg cells, we evaluated the FOXP3 expression and found the FOXP3 expression to be between 80–95%. When evaluating the level of CD4+CD127+CD25neg-int T cells, the cells were gated on CD4+ lymphocytes then on CD127+ and CD25neg-int.
Statistical analysis
This single-institution phase-II study was approved by the Ethics Committee of Funen County. The primary objective of the study was to determine response rate of the study treatment according to RECIST, whereas estimation of time to progression, overall survival and evaluation of tolerability of the regimen were secondary objectives. Patients were accrued to this study using a two-stage Simon's design [23] in order to improve the probability of observing the true response rate. Thus, if there were three or more responders among the first 21 evaluable patients, the accrual would be extended to a total of 40 evaluable patients with five patients per responder with an estimated response rate of 25%. Descriptive statistics were used to describe baseline patient characteristics, the cerebral and extra-cerebral objective response rate and toxicity. Time to progression and death were estimated using the Kaplan-Meier method, with overall survival measured from study entry until death from any cause. The T-cell data were analysed using a student t test.
Results
Patient characteristics
A total of 43 patients were enrolled in the study between June 2005 and
February 2007. Three patients did not meet protocol eligibility requirements
and were excluded from all analyses. Baseline patient and disease
characteristics for 40 eligible patients (21 men, 19 women) are shown in Table 1. Thirty-nine
patients were evaluable for toxicity as one patient died shortly after the
start of treatment. Median age of the patients was 55 years (range: 29–76
years), 16 patients were in WHO PS: 0 and 24 in PS: 1. All patients had non-resectable
brain metastases in progression: 15 had minor neurologic symptoms and were
treated with concomitant steroids. The remaining 25 patients had asymptomatic
brain metastases all found during screening procedures prior to planned
IL-2-based therapy. Among 40 patients, 34 had known primary cutaneous melanoma,
six had unknown primary melanoma. Thirty-nine patients had concomitant
extensive extra-cranial disease with 15% having liver metastases and 57% having
lung metastases. Before study entry, four patients had received whole-brain
radiotherapy, one had received stereotactic radiotherapy and three had
undergone surgery for brain metastases. Seven patients had received prior
immunotherapy and none prior cytotoxic therapy.
The patients completed a median of three cycles of therapy (range: 1–10
cycles). Eight patients received only one cycle because of early clinical
disease progression. Twenty-seven patients completed three cycles and were
evaluated for efficacy.
Table 1: Patient characteristics (n = 40)

Analysis of lymphopenia
All 40 patients were analysed for lymphopenia. Eight patients experienced no lymphopenia during treatment; five patients experienced grade 1; eight patients grade 2; 16 patients grade 3; and eventually three patients experienced grade 4 lymphopenia (< 0.2 x 109/l ). The observed lymphopenia rate was 68%.
Analysis of T-cell populations
A total of 12 patients were analysed for the presence of CD4+CD25+FOXP3+ Treg cells. None of the patients included in the analysis had objective response during treatment with THA and TMZ (eight patients had PD and four patients had SD). The percentage of Treg cells out of CD4 T cells was increased after treatment with TMZ and THA (p=0.001) (Figure 1A), while the total number of Treg cells was unchanged after treatment (p=0.8) (Figure 1B). The percentage of CD4 T cells out of lymphocytes (p=0.03) as well as the total number of CD4 T cells (p=0.04) decreased during treatment (Figure 2A and B). Furthermore, a decreased percentage of CD4+CD127+CD25neg-int T cells out of CD4 T cells (p=0.0001) along with a tendency to a decreased number of total CD4+CD127+CD25neg-int T-cells were observed (Figure 3). However, the latter did not reach statistical significance (p=0.2).
Toxicity
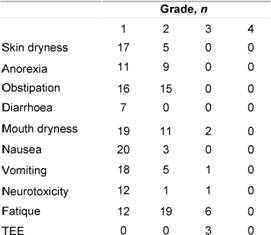
The reported non-haematologic adverse events
are summarized in Table 2.
The toxicity was primarily grade 1–2 with fatique, constipation and mouth
dryness. Four had thromboembolic events (TEE) all grade 3, which did not result
in discontinuation of therapy. These patients were treated with
anti-coagulants. Except for one patient with pancytopenia grade 3, no
significant neutropenia or thrombocytopenia and no grade 4 or treatment-related
deaths was observed. Thirteen patients were treated with antibiotics because of
lymphopenia. Among these, three patients had opportunistic infections or other
infections indicative of T-cell dysfunction with one patient having systemic
infection with Candida Albicans in the lungs and two patients with Legionella.
During the study, only two patients discontinued study treatment because of
toxicity. One patient discontinued because of grade 3 nausea and vomiting,
possibly related to the study drug and one patient because of grade 3
haematologic toxicity. Disease progression was the most common reason for
treatment discontinuation (62% of cases).
Table 2: Toxicity profile in 39 evaluable patients
Response
Objective response rates according to RECIST in the brain and outside CNS are shown in Table 3. Among the 40 patients, one patient obtained a CR in the brain, five a PR and ten a SD. Outside CNS, two patients obtained a CR, four a PR and ten a SD. One patient obtained a CR overall, while six patients had PR, leading to an overall response rate of 17.5%. Unfortunately, planned confirmation of response according to RECIST was not a part of our protocol; therefore, only four out of seven responses were confirmed after three months. A further eight patients had SD in both CNS and outside CNS, leading to a clinical benefit rate of 37.5%. The three patients referred to as non-evaluable for response outside the CNS were all patients with metastases in the brain as the only metastatic site. Among the seven patients who had an overall objective response, three were treated with concomitant steroids, four had symptomatic and three had asymptomatic brain metastases.
Table 3: Response in 40 patients
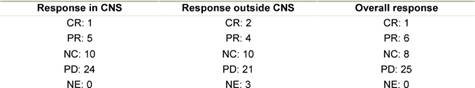
Table 4: Significant correlation between response and lymphocyte count p2=0.01
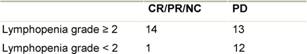
The correlation between lymphocyte count and response to treatment is shown in Table 4. Patients
with SD have been categorized together with patients obtaining an objective
response. We find a statistically significant correlation between efficacy and
lymphopenia (Fishers exact test, p= 0.01). The Kaplan-Meier estimate of time to
progression is shown in Figure
4. Median time to progression was 2.7 months (95% conf lim: 0.3–9.3
months). Kaplan-Meier estimate of overall survival is shown in Figure 5. The median survival time was
4.2 months (95% conf lim: 0.8–14.0 months). No difference in survival comparing
patients with symptomatic (n=15) and asymptomatic (n=25) brain
metastases was found (data not shown).
Figure 4: Kaplan-Meier estimate of time to progression in 40 patients. Median time to progression is 2.7 months (95% conf lim: 0.3–9.3 mths)
|
|
|
|
|
|
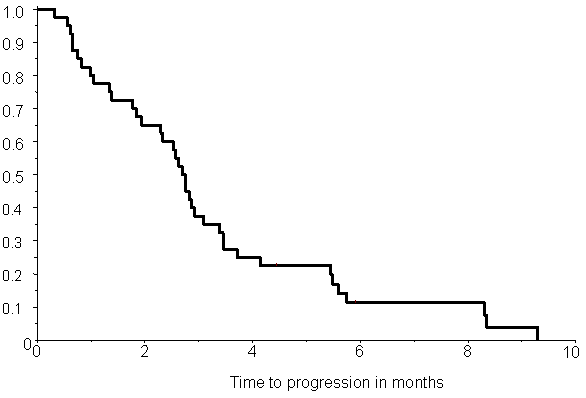
Figure 5: Kaplan-Meier estimate of overall survival in 40 patients. Median survival time is 4.2 months (95% conf lim: 0.8–14.0 months)

Discussion
In this phase II trial, we have investigated the combination of THA and a dose-intensified schedule of TMZ 150 mg/m2 seven days on/seven days off, in the treatment of malignant melanoma with brain metastases. In our study, we observed a response rate of 17.5% and a clinical benefit rate (CR + PR + SD) of 37.5%. The response rate using the classical schedule of TMZ single drug leads to a response rate of 6–9% [24,25], with further 10% obtaining SD. These two studies were the first to demonstrate any anti-tumour activity of TMZ in brain metastases of melanoma patients. A phase-II study by Schadendorf et al [26] using dose-intensified bi-weekly TMZ did not lead to significant response in brain metastases or extra-cerebral metastatic lesions (under 5%) [26]. A small study with extended dose of TMZ (six weeks, low dose) combined with THA showed a response rate of 12% and a clinical benefit of up to 31% in 26 patients [19,27]. Our study is the first phase-II study published combining the cyclic (seven days on/seven days off) regimen of TMZ and THA for patients with malignant melanoma suffering from brain metastases, showing a meaningful response rate.
Su et al were the first to report lymphopenia and opportunistic infections as an unexpected toxicity of extended-dose regimens of TMZ in melanoma patients [8]. They reported their retrospective experience with patients receiving this regimen alone or combined with either THA or low-dose interferon-alpha. Lymphopenia was not more common in patients treated with TMZ combined with THA, compared to TMZ alone. CD4+ lymphopenia was seen in 60% of patients, leading to opportunistic infections as well as other infections indicative of T-cell dysfunction. They concluded that Pneumocystis pneumonia prophylaxis should be considered for patients who develop sustained lymphopenia on TMZ. We can conclude the same from our study. We observed grade 2–4 lymphopenia during treatment in 27 of the 40 patients (67%). Interestingly, in this subgroup of patients significantly more responders were seen, indicating that immune parameters might influence the response of this combination regimen. From the study of Su et al [8], analysis by flow cytometry showed that the absolute CD4+ T-cell counts were depressed and that the CD4+CD25+ T-cell subset in some patients was markedly depressed, indicating that the lymphopenic effect of TMZ is relatively specific for the CD4+ subset.
Lately, it has become clear that Treg cells might play a role in inhibiting beneficial anti-tumour immune responses [28]. Treatment of cancer patients with low doses of cyclophosphamide has resulted in the depletion of Treg cells, which in theory might improve anti-tumour immunity [28,29]. Thus, we evaluated the impact of TMZ and THA on the Treg cell populations to determine whether the treatment had any effect on these cells. When analysing the presence of CD4+CD25+FOXP3+ Tregs in patients treated with TMZ and THA, we found that the percentage of Treg cells out of CD4 T cells was increased after treatment. However, this increase in Treg cell proportion did not result in an increased total number of Treg cells, since the percentage of CD4 T cells after treatment was lowered. Others have observed a preferential lymphopenia in the CD4+ compartment in melanoma patients after treatment with TMZ8. In the present study, we also observed a lowered percentage of CD4+ T cells out of total lymphocytes along with a lowered total number of CD4+ T cells after treatment with TMZ and THA. This indicates that other subsets of CD4+ T cells apart from the Treg cell subset are sensitive to the treatment. Interestingly, when evaluating the fraction of CD4+CD127+CD25neg-int out of CD4+ T cells, we found that this was lowered. Furthermore, we observed a tendency towards a lower number of total CD4+CD127+CD25neg-int T cells after treatment, suggesting that these cells were influenced by TMZ. The expression of CD127 is down regulated on activated effector CD4 T cells [30], as well as on Treg cells [31,32], thus it seems that TMZ affects the CD127+CD25neg-int subset, which mostly contains conventional naïve T cells [33] and memory T cells [34]. Unfortunately, we only obtained data from patients with PD and SD. Thus, we are unable to report whether the CD4 T cell subsets fluctuate in a similar way in patients responding to the treatment with TMZ and THA or if they change at all. This will be evaluated in an ongoing trial.
The TMZ and THA treatment was well tolerated with mainly grade 1–2 non-haematological toxicity and no significant haematological toxicity. However, the combination had significantly more TEE than either Thalidomide or TMZ alone [18,35]. It has previously been shown in multiple myeloma that, when THA is combined with cytotoxic agents or with steroids, the frequency of TEE increases [36]. Furthermore, it is well known that cancer in itself is associated with a higher risk of TEE [37]. This was the case in the study performed by CALGB [24], which was suspended and subsequently closed because of severe TEE and limited number of objective responses. In the CALGB study, THA was given in doses up to 400 mg daily. The lower dose of THA in our study may explain the more manageable TEEs in four patients having deep-vein thrombosis and none of the patients developing pulmonary embolism. No significance can be established because of the small number of patients; however, in future studies one should be aware of this complication associated with this treatment combination and should maybe use lower doses of THA.
We conclude that the combination of TMZ, using the bi-weekly dose-intense schedule and THA 100–200 mg/d, is a safe regimen, leading to clinical efficacy in patients with brain metastases from malignant melanoma. Most importantly, it seems that patients who develop lymphopenia during therapy have a higher chance of obtaining objective response. The potential immunologic mechanism behind this will be the subject of future investigations, focusing on the potential benefit of Treg down regulation. Further evaluations using larger patient numbers and including different therapy schedules will be considered for the future. Concerning the schedule of TMZ, we await the ongoing EORTC 18032 phase-III trial, investigating the bi-weekly regimen of TMZ versus standard Dacarbazine in previously untreated patients with metastatic melanoma.
References
1. Lee ML, Tomsu K and Von Eschen KB (2000) Duration of survival for disseminated malignant melanoma: results of a meta-analysis Melanoma Res 10 1 81–92 PMID 10711644
2. Tarhini AA and Agarwala SS (2004) Management of brain metastases in patients with melanoma Curr Opin Oncol 16 2 161–6 PMID 15075910 doi: 10.1097/00001622-200403000-00014
3. Meier S, Baumert BG, Maier T et al (2004) Survival and prognostic factors in patients with brain metastases from malignant melanoma Onkologie 27 2 145–9 PMID 15138346 doi: 10.1159/000076903
4. Sampson JH, Carter JH, Friedman AH et al (1998) Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma J Neurosurg 88 1 11–20 PMID 9420067 doi: 10.3171/jns.1998.88.1.0011
5. Payne MJ, Pratap SE and Middleton MR (2005) Temozolomide in the treatment of solid tumours: current results and rationale for dosing/scheduling Crit Rev Oncol Hematol 53 3 241–52 PMID 15718149 doi: 10.1016/j.critrevonc.2004.10.004
6. Brock CS, Newlands ES, Wedge SR et al (1998) Phase I Trial of Temozolomide Using an Extended Continuous Oral Schedule Cancer Res 58 19 4363–7 PMID 9766665
7. Tolcher AW, Gerson SL, Denis L et al (2003) Marked inactivation of O6-alkylguanine-DNA alkyltransferase activity with protracted temozolomide schedules Br J Cancer 88 7 1004–11 PMID 12671695 doi:10.1038/sj.bjc.6600827
8. Su YB, Sohn S, Krown SE, Livingston PO, Wolchok JD, Quinn C, Foster T, Sepkowitz KA and Chapman PB (2004) Selective CD4 +lymphopenia in melanoma patients treated with temozolomide: A toxicity with therapeutic implications J Clin Oncol 22 4 610–6 PMID 14726505 doi:10.1200/JCO.2004.07.060
9. Rietschel P, Wolchok JD, Krown S et al (2008) Phase II Study of Extended-Dose Temozolomide in Patients With Melanoma J Clin Oncol 26 14 2299–304 PMID 18467721 doi: 10.1200/JCO.2007.14.5292
10. Thomas DA and Kantarjian HM (2000) Current role of thalidomide in cancer treatment Curr Opin Oncology 12 6 564–73 PMID 11085456 doi:10.1097/00001622-200011000-00009
11. D'Amato RJ, Loughnan MS, Flynn E et al (1994) Thalidomide is an Inhibitor of Angiogenesis Proc Natl Acad Sci USA 91 9 4082–5 PMID 7513432 doi:10.1073/pnas.91.9.4082
12. Rajkumar SV (2001) Thalidomide in the treatment of plasma cell malignancies J Clinical Oncol 19 16 3593–5 PMID 11504740
13. Moreira AL, Sampaio EP, Zmuidzinas A et al (1993) Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation J Exp Med 177 6 1675–80 PMID 8496685 doi:10.1084/jem.177.6.1675
14. Meierhofer C and Wiedermann CJ (2003) New insights into the pharmacological and toxicological effects of Thalidomide Curr Opin Drug Discov Devel 6 1 92–9 PMID 12613280
15. Meierhofer C, Dunzendorfer S and Wiedermann CJ (2001) Theoretical Basis for the Activity of Thalidomide BioDrugs 15 10 681–703 PMID 11604049 doi:10.2165/00063030-200115100-00005
16. Franks ME, MacPherson GR and Figg WD (2004) Thalidomide Lancet 363 9423 1802–11 PMID 15172781 doi:10.1016/S0140-6736(04)16308-3
17. Kumar S, Witzig TE and Rajkumar SV (2004) Thalidomide: Current role in the treatment of non-plasma cell malignancies J Clin Oncol 22 12 2477–88 PMID 15197211 doi:10.1200/JCO.2004.10.127
18. Vestermark LW, Larsen SU, Lindeløv B and Bastholt L (2008) A phase II study of Thalidomide in patients with brain metastases from malignant melanoma Acta Oncologica 47 8 1526–30 PMID 18607876 doi: 10.1080/02841860801918521
19. Hwu WJ, Lis E, Menell JH et al (2005) Temozolomide Plus Thalidomide in Patients with Brain Metastases from Melanoma Cancer 103 12 2590–7 PMID 15861414 doi:10.1002/cncr.21081
20. Zeldis JB, Williams BA, Thomas SD et al (1999) STEPS: a comprehensive program for controlling and monitoring access to thalidomide Clin Ther 21 2 319–30 PMID 10211535 doi: 10.1016/S0149-2918(00)88289-2
21. Trotti A, Colevas AD, Setser A et al (2003) CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment Seminars in Rad Oncol 13 3 176–81 PMID 12903007 doi:10.1016/S1053-4296(03)00031-6
22. Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors J Natl Cancer Inst 92 3 205–16 PMID 10655437 doi:10.1093/jnci/92.3.205
23. Simon R (1989) Optimal two-stage designs for phase II clinical trials Control Clin Trials 10 1 1–10 PMID 2702835 doi:10.1016/0197-2456(89)90015-9
24. Krown SE, Niedzwiecki D, Hwu W-J et al (2006) Phase II study of temozolomide and thalidomide in patients with metastatic melanoma in the brain : High rate of thromboembolic events (CALGB 500102) Cancer 107 8 1883–90 PMID 16986123 doi:10.1002/cncr.22239
25. Boogerd W, de Gast GC and Dalesio O (2006) Temozolomide in advanced Malignant Melanoma with smal brain metastases. Can We Withhold cranial irradiation? Cancer 109 2 306–12 PMID 17149755 doi:10.1002/cncr.22411
26. Schadendorf D, Hauschild A, Ugurel S et al (2006) Dose-intensified bi-weekly temozolomide in patients with asymptomatic brain metastases from malignant melanoma: a phase II DeCOG/ADO study Ann Oncol 17 10 1592–7 PMID 17005632 doi:10.1093/annonc/mdl148
27. Laber D, Okeke R, Arce-Lara C et al (2006) A phase II study of extended dose temozolomide and thalidomide in previously treated patients with metastatic melanoma J Cancer Res Clin Oncol 132 9 611–6 PMID 16741726 doi:10.1007/s00432-006-0114-8
28. Zou W (2006) Regulatory T cells, tumour immunity and immunotherapy Nat Rev Immunol 6 4 295–307 PMID 16557261 doi: 10.1038/nri1806
29. Ghiringhelli F, Menard C, Puig P et al (2007) Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients Cancer Immunol Immunother 56 5 641–8 PMID 16960692 doi:10.1007/s00262-006-0225-8
30. Lim HW and Kim CH (2007) Loss of IL-7 Receptor {alpha} on CD4+ T Cells Defines Terminally Differentiated B Cell-Helping Effector T Cells in a B Cell-Rich Lymphoid Tissue J Immunol 179 11 7448–56 PMID 18025189
31. Seddiki N, Santner-Nanan B, Martinson J et al (2006) Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells J Exp Med 203 7 1693–1700 PMID 16818676 doi:10.1084/jem.20060468
32. Liu W, Putnam AL, Xu-yu Z et al (2006) CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells J Exp Med 203 7 1701–11 PMID 16818678 doi:10.1084/jem.20060772
33. Fry TJ and Mackall CL (2005) The Many Faces of IL-7: From Lymphopoiesis to Peripheral T Cell Maintenance J Immunol 174 11 6571–6 PMID 15905493
34. Kondrack RM, Harbertson J, Tan JT et al (2003) Interleukin 7 Regulates the Survival and Generation of Memory CD4 Cells J Exp Med 198 12 1797–1806 PMID 14662907 doi:10.1084/jem.20030735
35. Agarwala SS, Kirkwood JM, Gore M et al (2004) Temozolomide for the Treatment of Brain Metastases Associated With Metastatic Melanoma: A Phase II Study J Clin Oncol 22 11 2101–7 PMID 15169796 doi:10.1200/JCO.2004.11.044
36. Zangari M, Anaissie E, Barlogie B et al (2001) Increased risk of deep-vein thrombosis in patients with multiple myeloma receiving thalidomide and chemotherapy Blood 98 5 1614–5 PMID 11520815 doi:10.1182/blood.V98.5.1614
37. Gerber DE, Grossman SA and Streiff MB (2006) Management of Venous Thromboembolism in Patients With Primary and Metastatic Brain Tumors J Clin Oncol 24 8 1310–8 PMID 16525187 doi:10.1200/JCO.2005.04.6656
Supplementary figures
Figure 1: Percentage and total number of Treg cells in
malignant melanoma patients before and after treatment with thalidomide and
temozolomide. MNCs were stained with PE anti-human FOXP3,
PerCP anti-human CD4, APC anti-human CD25. For defining the population of Treg
cells, the cells were first gated on CD4+ lymphocytes then on CD4+
and CD25high. On the pre-sample, the level of CD4+CD25high
out of CD4+ T cells was fixed at 2% and the following two samples
(day 14 and day 28) were analysed on the same day using the same gates. To
confirm that the cells within the gate were Treg cells, we evaluated
the FOXP3 expression and found the FOXP3 expression to be between 80%-95%. (A)
Percentage of Treg cells out of CD4 T cells. (B) Total number of Treg
cells/µl blood
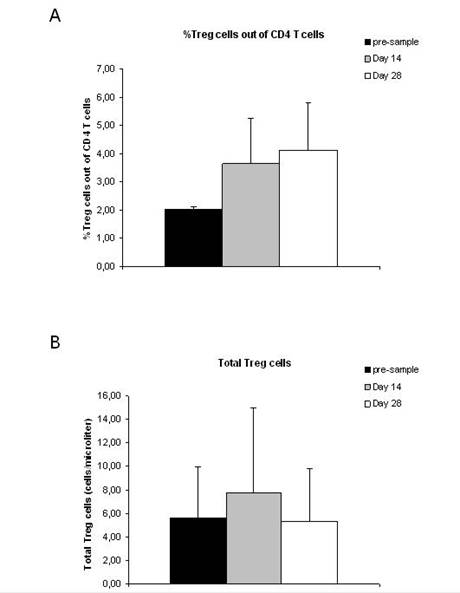
Figure 2: Percentage and total number of CD4 T cells in malignant
melanoma patients before and after treatment with thalidomide and temozolomide.
(A) Percentage of CD4 T cells out of lymphocytes. (B) Total number of CD4 T
cells
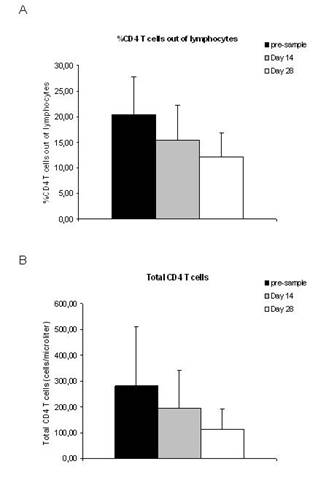
Figure 3: Percentage and total number of CD4+CD25neg-intCD127+
T cells in malignant melanoma patients before and after treatment with
thalidomide and temozolomide.
MNCs were stained with FITC anti-human CD127, PerCP anti-human CD4, APC
anti-human CD25. When evaluating the level of CD4+CD127+CD25neg-int
T cells, the cells were gated on CD4+ lymphocytes then on CD127+
and CD25neg-int