Metastatic pheochromocytoma and paraganglioma: a retrospective multicentre analysis on prognostic and predictive factors to chemotherapy
Samara T Pacheco1, Mauro D Donadio1, Felipe Almeida1, Juan M O’Connor3, Valeria de Miguel2, Mariano Dioca4, Jose Huaman7, Arinilda C Bragagnoli5, Rui F Weschenfelder6, Paola M Beltran7 and Rachel P Riechelmann1
1AC Camargo Cancer Center, São Paulo, Brazil
2Hospital Italiano de Buenos Aires, Buenos Aires, Argentina
3Instituto Alexander Fleming, Buenos Aires, Argentina
4Instituto de Oncologia Ángel H. Roffo, Buenos Aires, Argentina
5Hospital de Amor, Barretos, Brazil
6Hospital Moinho de Ventos, Porto Alegre, Brazil
7Instituto Nacional Enfermidades Neoplasicas, Lima, Peru
Abstract
Background: Prognostic and predictive markers in metastatic pheochromocytoma and paraganglioma (mPPGL) are unknown. We aimed to evaluate epidemiology of mPPGL, and prognostic factors of overall survival (OS) and predictive markers of treatment duration with first-line chemotherapy (TD1L).
Patients and methods: Retrospective multicentre study of adult patients with mPPGL treated in Latin American centres between 1982 and 2021.
Results: Fifty-eight patients were included: 53.4% were female, median age at diagnosis of mPPGL was 36 years and 12.1% had a family history of PPGL. The primary site was adrenal, non-adrenal infradiaphragmatic and supradiaphragmatic in 37.9%, 34.5% and 27.6%, respectively. 65.5% had a functioning tumour and 62.1% had metachronous metastases. Positive uptakes were found in 32 (55.2%) 68Gallium positron emission tomography (PET/CT), 27 (46.6%) 2-deoxy-2-[fluorine-18]fluoro-D-glucose PET/CT and 37 (63.8%) of 131Iodine-metaiodobenzylguanidine (MIBG) tests. Twenty-three (40%) patients received first-line chemotherapy, with cyclophosphamide, vincristine and dacarbazine used in 12 (52%) of patients. At a median follow-up of 62.8 months, median TD1L was 12.8 months. Either functional exams, tumour functionality, pathological characteristics or primary tumour location were significantly associated with response or survival. Yet, negative MIBG, Ki67 ≥ 10%, infradiaphragmatic location and functional tumours were associated with numerically inferior OS.
Conclusions: In patients with mPPGL, prognostic and predictive factors to chemotherapy are still unknown, but negative MIBG uptake, Ki67 ≥ 10%, infradiaphragmatic location and functional tumours were numerically linked to worse OS. Our results should be further validated in larger and independent cohorts.
Keywords: pheochromocytoma, paraganglioma, prognostic, predictive, survival, chemotherapy, metastasis
Correspondence to: Rachel P Riechelmann
Email: rachel.riechelmann@accamargo.org.br
Published: 20/03/2023
Received: 17/11/2022
Publication costs for this article were supported by ecancer (UK Charity number 1176307).
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Paragangliomas and pheochromocytomas (PPGL) are neuroendocrine neoplasms that arise from paraganglia and chromaffin cells but of the adrenal medulla, respectively. PPGL are rare, with estimated incidence of about 0.6 cases per 100,000 people/year [1], and both can secrete catecholamines and neuropeptides, with resulting paroxysmal hypertension, constipation, episodic headache, sweating and pallor, tremors and palpitations [2]. To date, there are no established clinical, histopathological or biochemical features to determine metastatic behaviour. Hence, a diagnosis of malignant PPGL relies on the presence of metastases, which occurs in approximately 10%–20% of cases, with heterogeneous disease course [1, 3]. In an attempt to evaluate prognostic factors for malignant PPGL, a multiparameter score (PASS) was conceived for adrenal gland pheochromocytoma based on the evaluation of morphologic features of tumour cells. The PASS score, published in 2002, considered high risk of malignant behaviour when the score was ≥4 [4]. In 2014, a new score was developed, the Grading of Adrenal Pheochromocytoma and Paraganglioma (GAPP) scoring system, which added the immunohistochemistry (IHC) Ki67 index and the presence of catecholamine secretion to the PASS parameters. The score can range from 0 to 10, higher scores being linked to higher risk of metastases [5]. A recent study evaluated the accuracy of PASS, GAPP and a modified GAPP score, where investigators also evaluated the lack of succinate dehydrogenase complex iron sulphur subunit B gene (SDHB) IHC expression as prognostic factor for metastasis in 72 PPGL cases [6, 7]. Both GAPP scores and modified version were more accurate than PASS. Nevertheless, the reproducibility of these scores is variable because of interobserver variations [4].
While prognostic markers of malignant potential in PPGL have been studied, little is known about prognostic factors for survival and/or predictive markers of benefit from systemic therapies, even for the most commonly used first-line systemic treatment, the combination chemotherapy with cyclophosphamide, vincristine and dacarbazine (CVD). The effects of this combination were assessed in a systematic review and meta-analysis including 50 patients, showing a complete or partial response rate in 4% and 37%, respectively, and stable disease in 14% of patients [13]. No prognostic or predictive factors, however, were assessed.
At present, data on the efficacy of tyrosine kinase inhibitors in metastatic paraganglioma/pheochromocytoma are derived from phase II studies [14–16]. Sunitinib was evaluated in a European randomised, placebo-controlled trial phase II trial with malignant PPGL (FIRST-MAPPP trial). Median progression-free survival (PFS) was 8.9 versus 3.6 months [17]. In addition to chemotherapy and tyrosine kinase inhibitor (TKI), the use of nuclear medicine in treatment for metastatic PPGL (mPPGL) is well established. A meta-analysis was performed which showed a complete or partial tumour response in 3% and 27% of patients treated with 131Iodine-metaiodobenzylguanidine (MIBG), respectively. In addition, 52% of patients had stable disease; [18] but given the often indolent nature of pheochromocytoma and metastatic paraganglioma, it is unclear whether this is due to treatment effects or the natural history of the disease [19]. As an alternative to MIBG treatment, if the tumour is a somatostatin receptor positive upon imaging, peptide receptor radionuclide therapy (PRRT) or somatostatin analogues may be considered. A meta-analysis of 201 patients with mPPGL showed that PRRT achieved an objective response rate of 25% and a disease control rate of 84%. Clinical and biochemical responses were seen in 61% and 64% of the patients, respectively [20]. Beyond the first-line setting, temozolomide has been described as a potential therapeutic tool, mainly in the second line [21, 22].
Yet, some patients with mPPGL have indolent disease, with disease stabilisation for years, and some patients present aggressive and rapidly progressing tumours; while some patients have highly responsive tumours to chemotherapy or sunitinib, others present upfront refractory disease. Thus, prognostic and predictive markers in mPPGL are unclear. Our primary objective was to evaluate prognostic factors of overall survival (OS) and predictive markers of treatment duration with first-line chemotherapy for patients with mPPGL. Our secondary objective was to describe the epidemiology of mPPGL patients.
Materials and methods
This was a retrospective, multicentre study of patients with mPPGL, defined by radiological exams. Consecutive patients from the seven following hospitals in Latin America specialised in cancer treatment were included: AC Camargo Cancer Center (São Paulo, Brazil), Hospital de Amor (Barretos, Brazil), Hospital Moinho de Ventos (Porto Alegre, Brazil), Instituto Nacional Enfermidades Neoplasicas (Lima, Peru), Hospital Italiano de Buenos Aires (Buenos Aires, Argentina), Instituto Alexander Fleming (Buenos Aires, Argentina), Instituto de Oncologia Ángel H. Roffo (Buenos Aires, Argentina). The protocol was approved by the Ethics Committee Boards of participating institutions. Patients over 16 years old diagnosed with histologically confirmed PPGL and radiologically documented synchronous or metachronous metastases were eligible. Cases seen only once, as second opinions, where follow-up information was not available, were excluded. Consecutive cases were selected from centres’ databases from January 1982 through September 2021, using the C47 or C74 coding of the International Classification of Diseases, 11th Revision.
The following data were collected from medical charts: gender, date of birth and date of initial diagnosis and of metastatic disease, country of origin, family history of PPGL, known genetic syndrome, smoking status, Eastern Cooperative Oncology Group (ECOG) performance status at the time of mPPGL, time interval from initial diagnosis until evidence of metastases in initially non-stage IV cases, metastatic sites, primary site location, presence of functioning syndrome, functioning imaging tests (68Gallium positron emission tomography (68Ga-PET/CT), 2-deoxy-2-[fluorine-18]fluoro-D-glucose PET/CT (18F-FDG-PET/CT), MIBG scintigraphy), Ki67 IHC expression index, type and lines of treatments, date of progressions, survival status on last follow-up and date of last follow-up.
Patients from AC Camargo Cancer Center with archived paraffin-embedded tumour tissues available had their pathology material revised and evaluated for the IHC expression of SDHB in a tissue microarray by a pathologist with expertise in the diagnosis of PPGL (FA).
Our main objective was to investigate prognostic and predictive factors to systemic chemotherapy, the most common regimen being utilised for mPPGL patients in our region. To increase internal validity, we limited our analytical sample to patients with mPPGL who received systemic chemotherapy in first-line. The primary endpoint was OS, which was calculated from the date of radiological documentation of metastatic disease to the date of death of any cause. The main secondary endpoint was treatment duration of disease control in first-line (DDC1L), defined as the time from day 1 cycle 1 (D1C1) of first-line chemotherapy to D1C1 of second-line treatment; patients who died before the receipt of second-line treatment, regardless of cause, were censored on the date of death. DDC1L was chosen as the main outcome variable to investigate predictive factors because we assume it is a proxy of PFS, and because we could not retrieve radiological exams for proper RECIST determination. Response to first-line chemotherapy, as documented in medical charts, was explored as an outcome variable, with patients being categorised into two groups: complete or partial response, and stable disease or upfront tumour progression.
Prognostic and predictive variables evaluated for association with OS and DDC1L were: timing of metastases (metachronous or synchronous), positive metastatic uptake on 18F-FDG-PET/CT, on 68Ga-PET/CT or on MIBG, tumour functionality, primary tumour location (infradiaphragmatic or supradiaphragmatic) and Ki67 cut-off values (≥3% or ≥10%). Synchronous metastases were defined when detected within 6 months of the initial diagnosis of localised PPGL. For the analysis of predictive and prognostic factors, we considered a Ki67 cut-off point of 3%, according to extrapolation of the GAPP score data, in which values above this level are encountered in moderately or poorly differentiated tumours. As an exploratory analysis, we also evaluated the prognostic and predictive effects of Ki67 cut-off of 10%, based on data from patients with well-differentiated gastro-entero-pancreatic neuroendocrine tumours treated with targeted agents [8]. The Ki67 index captured was preferably that from metastases, if both primary and metastatic lesions were available.
Descriptive statistics were used to summarise absolute and relative frequencies and medians. DDC1L and OS were estimated by Kaplan–Meier method. The reverse Kaplan–Meier method was used to estimate the follow-up time. The unadjusted log-rank test was used to compare OS and DDC1L times according to prognostic and predictive variables, respectively. To determine the association between tumour response and predictive variables, we used the chi-square or exact test of Fisher. For all analyses, two-tailed p value < 0.05 was deemed significant. All analyses were performed in SPSS version 24.
Results
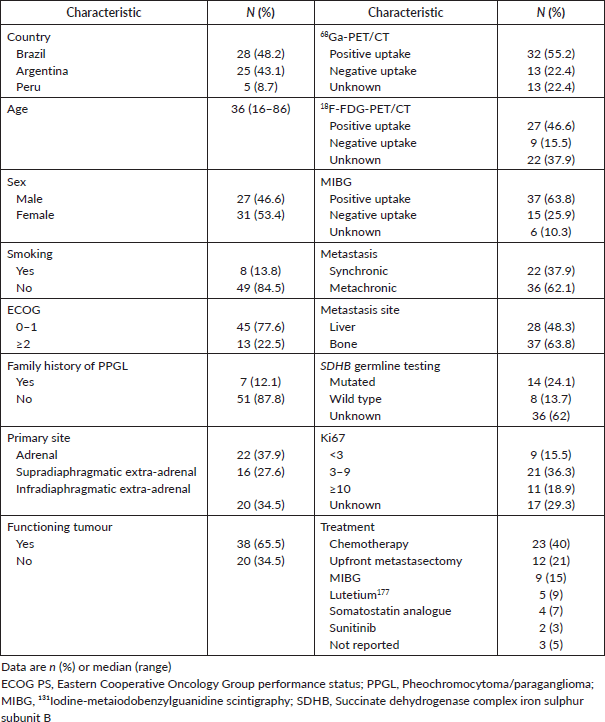
From 1980 to 2018, 58 patients with mPPGL were identified and included: 31 (53.4%) were female, median age at diagnosis of metastatic disease was 36 years (16–86), and seven (12.1%) patients had a family history of PPGL. The primary site was adrenal, non-adrenal infradiaphragmatic and supradiaphragmatic in 37.9%, 34.5% and 27.6% of cases, respectively. Thirty-eight (65.5%) patients had a functioning tumour and 62.1% had metachronous metastases. The great majority (84.9%) of patients had a 68Ga-PET/CT at some point during the course of their mPPGL. An 18F-FDG-PET/CT was performed by 36 (62%) and an MIBG was performed in 92.8% of the patients. Among these cases, positive uptakes were found in 32 (55.2%) 68Ga-PET/CT, in 27 (46.6%) 18F-FDG-PET/CT and in 37 (63.8%) of MIBG tests. Table 1 summarises the characteristics of patients.
Twenty-three (40%) patients received first-line chemotherapy and comprise the population for the evaluation of prognostic and predictive factors. Other directed therapies administered in first-line were upfront metastasectomy in 12 (21%) patients, therapeutic MIBG in 9 (15%), Lutetium177 in 5 (9%), a somatostatin analogue in 4 (7 %) and sunitinib in 2 (3%) cases.
Table 1. Baseline characteristics – general population (all = 58).
Prognostic and predictive factors in first-line chemotherapy in mPPGL
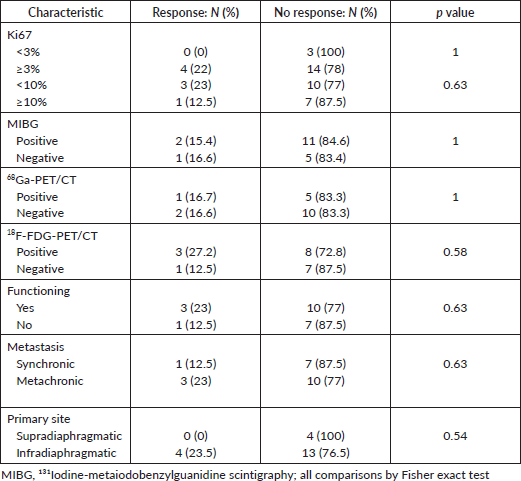
Table 2 depicts the characteristics of 23 patients treated with first-line chemotherapy. The median age was 43 years (16–71). The most commonly used chemotherapy regimen was CVD in nearly half (12; 52%) of patients. Of 21 patients with radiological response assessment, 4 (19%) achieved a partial response, 10 (47.6%) had stable disease and 7 (33.3%) had disease progression as the best response. Either tumour uptake on MIBG (p = 1.0), 68Ga-PET/CT (p = 1.0) or 18F-FDG-PET/CT (0.58), the Ki67 index (cut off 3% (p = 1) or 10% (p = 0.63)), tumour functionality (p = 0.63), timing of metastases (p = 0.63) or primary tumour location (p = 0.54) influenced the response to first-line chemotherapy (Table 3).
Table 2. Baseline characteristics – patients treated with first-line chemotherapy (N = 23).
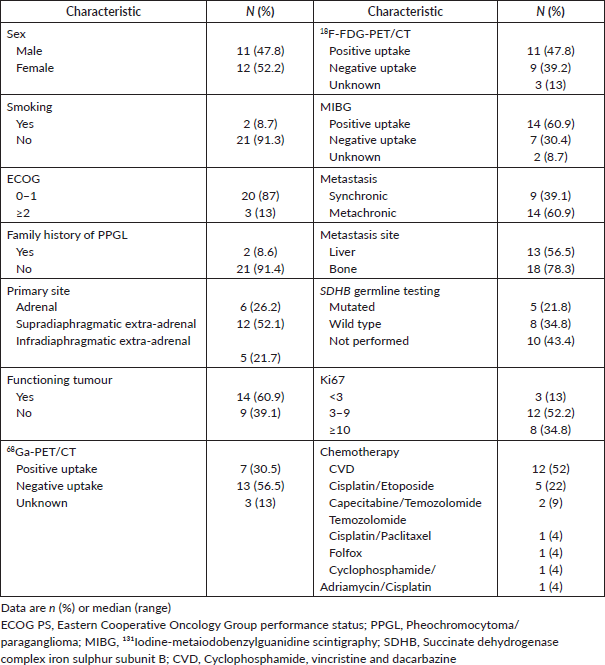
Table 3. Predictive factors to first-line chemotherapy.
At a median follow-up of 62.8 months, the median DDC1L was 12.8 months. During follow-up, 13 patients passed away and the median OS of all 23 patients was 71.3 months. No variable was significantly prognostic for OS or predictive of DDC1L. Non-significant large numerical differences were observed in DDC1L according to Ki67 (Ki67 < 10%: median of 13.7 months versus ≥10%: 1.8 months; p = 0.1; Figure 1) and OS according to MIBG uptake (median of 72.6 for positive uptake versus 42.1 months for negative uptake; p = 0.13; Figure 2) by primary tumour location (supradiaphragmatic location: median of 221 × 71.3 months for adrenal and infradiaphragmatic; p = 0.28; Figure 3); or to tumour functionality (non-functioning tumours: median of 221 × 71.3 months; p = 0.39; Figure 4).
Discussion
While prognostic and predictive factors in mPPGL remain unknown, our data suggest that negative baseline MIBG uptake, Ki67 ≥ 10%, infradiaphragmatic location and functional tumours may be associated with inferior OS. Our multicentre study also reports similar epidemiology to other series of mPPGL.
Unfortunately, financial issues do not yet allow molecular and genetic analyses to be performed routinely for all patients with PPGL in developing countries. Less than 40% of our sample had SDHB mutation test, but the prevalence found of 24% is also consistent with studies that showed that SDH mutations prevalence estimated to lie between 10% and 30% of PPGL [9]. Only 12%, however, had a family history, which is described in between a quarter and one third of PPGL cases [7, 10, 11]. Although it may be associated with the difficulty of accessing high-quality records on family history, a possible cause for the lower frequency of family association in our country is the high miscegenation characteristic of Latin American countries, which can decrease the homozygosity of autosomal recessive syndromes, more associated with PPGL [12].
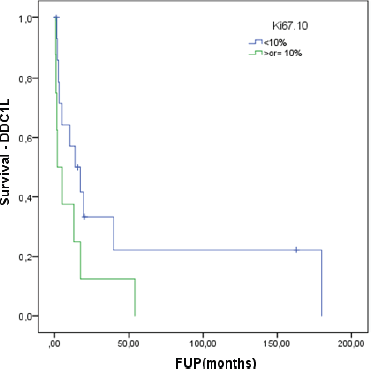
Figure 1. Time of treatment in first-line chemotherapy according to Ki67.
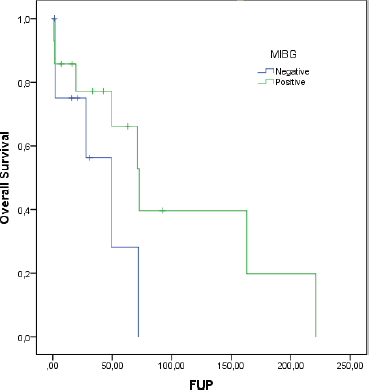
Figure 2. OS according to MIBG uptake.
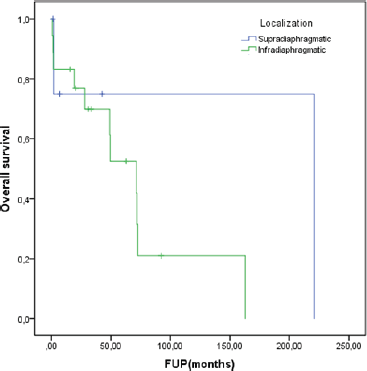
Figure 3. OS according to localisation.
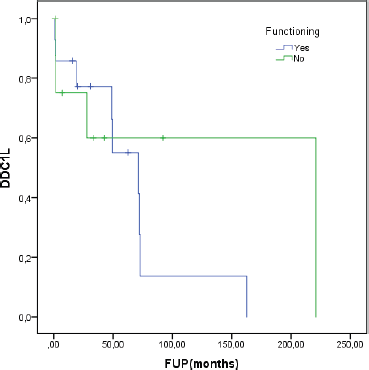
Figure 4. Disease control in first-line (DDC1L) according to functioning tumour.
Regarding functionality, about 60% of patients treated with chemotherapy had a functioning tumour, with positivity for MIBG and low Ki67 (<10%). Despite that, the main treatment used was chemotherapy and the regimens most used were CVD (52%) and cisplatin plus etoposide (22%). While CVD chemotherapy historically provides a partial response in 37% of patients, we found 19% of response and this lower rate may be associated with the predominance of low-grade tumours in our sample. Fifteen of the 23 patients treated with first-line chemotherapy had Ki67 less than 10%.
We could not identify, however, significant factors associated with DDC1L or response to first-line chemotherapy or prognostic factors of OS among these patients. Nevertheless, we observed a trend for lower DDC1L among patients whose tumours had a Ki67 ≥ 10%, and worse OS in patients with negative tumour uptake in MIBG, infradiaphragmatic/adrenal location or with functioning tumours. These findings point to a more aggressive behaviour in these subgroups and a larger number of patients could overcome a potentially underpowered analysis of our small sample.
Because of the rarity of mPPGL, very few studies have been undertaken to investigate factors associated with treatment benefits. Most of the subsequent studies concerning Ki67 in adrenal and/or extra-adrenal pheochromocytomas were aimed at demonstrating that an elevated proliferative activity was more indicative of malignant behaviour [23–27]. In localised PPGL, the best prognostic cut-off varies in studies: some had identified 3% [23], some authors 6% [25], others 5% [26, 27]. In the metastatic setting, although Ki67 does not have a well-defined role as a prognostic marker, in our study, the data suggest, as in neuroendocrine neoplasms of the gastro-entero-pancreatic tract, a worse survival in those with Ki67 > 10%.
In an early study, most pheochromocytomas (21 out of 29 patients) accumulated 18F-FDG-PET/CT [28]. FDG uptake was observed in a greater proportion of malignant than benign tumours, and in those tumours that did not accumulate MIBG, took up FDG. The discrepancy between MIBG and FDG uptake has since been observed in other studies especially in patients with metastatic disease [29, 30]. This was postulated to be due to tumour dedifferentiation and loss of specific cellular characteristics, such as cell membrane norepinephrine and vesicular monoamine transporter systems responsible for MIBG uptake [30]. Although not significant, our study suggests that patients with positive MIBG have a longer survival, inferring a higher level of cell differentiation and less aggressive disease.
Tumour functionality may also be associated with prognosis. Some mPPGL are associated with high morbidity and mortality secondary to hypersecretion of catecholamines and metanephrines leading to hypertension, cardiovascular disease and even death [8] and, in our study, we actually found data that suggest lower OS in functioning tumours. Similarly, the European retrospective MAPP-Prono Study evaluated 169 patients from 18 centres and found that better survival was associated with head and neck paraganglioma (almost never functional), age < 40 years, metanephrines less than fivefold the upper limits of the normal range and low proliferative index. In multivariate analysis, hypersecretion (HR: 3.02 (1.65–5.55); p = 0.0004) was identified as an independent significant prognostic factor of worse OS [31]. SDHB mutations were evaluated but it could not be confirmed as a major prognostic parameter in PPGL and suggest additional key molecular events involved in tumour progression.
Our study has several limitations that should be acknowledged. Being a retrospective study on a rare disease, we covered a period of 39 years, where heterogeneity of certain therapeutic managements has occurred. Although we planned to enroll all consecutive cases, selection bias is possible and we do not have the exact number of excluded cases based on potential missing files. Additionally, it was not possible to collect reliable information on treatment-related adverse events, dose intensity, biochemical response or details on radiological response. Furthermore, the retrospective design did not permit us to obtain complete information about Ki67 evaluation of all centres. The small number of the enrolled patients represents another limitation, which may prevent unveiling possible predictors and to perform adjusted analyses to identify independent predictors of DDC1L and OS, although the rarity of chromaffin tumours should be considered.
Future research in rare cancer populations is possible only through collaboration. Our study is an example of such an effort conducted by members of Latin Americans institutions. Global collaborative studies are pivotal steps for the oncology community to learn the biology, patient characteristics and treatment outcomes of rare cancers.
Conclusion
In patients with mPPGL, prognostic and predictive factors to first-line chemotherapy are still unknown. Non-significant findings suggest that negative MIBG uptake, Ki67 ≥ 10%, infradiaphragmatic location and functional tumours are associated with lower OS. Our results should be further validated in independent and larger cohorts.
Conflicts of interest
No conflicts of interest to declare.
Funding
This work received no funding.
References
1. Neumann H, Junior W, and Eng C (2019) Pheochromocytoma and paraganglioma N Engl J Med 381(6) 552–565 https://doi.org/10.1056/NEJMra1806651 PMID: 31390501
2. Gomez A, Soares D, and Costa A, et al (2019) Pheochromocytoma and paraganglioma: implications of germline mutation investigation for treatment, screening, and surveillance Arch Endocrinol Metab 63(4) 369–375 PMID: 31365623
3. Lam A (2017) Update on adrenal tumours in 2017 World Health Organization (WHO) of endocrine tumours Endocr Pathol 28 213–227 https://doi.org/10.1007/s12022-017-9484-5 PMID: 28477311
4. Thompson L (2002) Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases Am J Surg Pathol 26(5) 551–566 https://doi.org/10.1097/00000478-200205000-00002 PMID: 11979086
5. Kimura N, Takekoshi K, and Naruse M (2018) Risk stratification on pheochromocytoma and paraganglioma from laboratory and clinical medicine J Clin Med 7 242 https://doi.org/10.3390/jcm7090242 PMID: 30150569 PMCID: 6162838
6. Koh J, Ahn S, and Kim H, et al (2017) Validation of pathological grading systems for predicting metastatic potential in pheochromocytoma and paraganglioma PLoS One 12(11) e0187398 https://doi.org/10.1371/journal.pone.0187398 PMID: 29117221 PMCID: 5678867
7. Fishbein L (2016) Pheochromocytoma and paraganglioma: genetics, diagnosis, and treatment Hematol Oncol Clin North Am 30(1) 135–150 https://doi.org/10.1016/j.hoc.2015.09.006
8. Caplin M, Pavel M, and Ruszniewski P (2014) Lanreotide in metastatic enteropancreatic neuroendocrine tumors N Engl J Med 371(16) 1556–1557 https://doi.org/10.1056/NEJMoa1316158 PMID: 25317881
9. Schiavi F, Boedeker C, and Bausch B, et al (2005) Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene J Am Med Assoc 294(16) 2057–2063 https://doi.org/10.1001/jama.294.16.2057
10. Neumann H, Bausch B, and McWhinney S, et al (2002) Germ-line mutations in nonsyndromic pheochromocytoma N Engl J Med 346(19) 1459–1466 https://doi.org/10.1056/NEJMoa020152 PMID: 12000816
11. Erlic Z, Rybicki L, and Peczkowska M, et al (2009) Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients Clin Cancer Res 15(20) 6378–6385 https://doi.org/10.1158/1078-0432.CCR-09-1237 PMID: 19825962
12. Lenders J, Duh Q, and Eisenhofer G, et al (2014) Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline J Clin Endocrinol Metab 99(6) 1915–1942 https://doi.org/10.1210/jc.2014-1498 PMID: 24893135
13. Niemeijer N, Alblas G, and van Hulsteijn L, et al (2014) Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: systematic review and meta-analysis Clin Endocrinol (Oxf) 81 642–651 https://doi.org/10.1111/cen.12542 PMID: 25041164
14. Joshua A, Ezzat S, and Asa S, et al (2009) Rationale and evidence for sunitinib in the treatment of malignant paraganglioma/pheochromocytoma J Clin Endocrinol Metab 94 5–9 https://doi.org/10.1210/jc.2008-1836
15. Ayala-Ramirez M, Chougnet C, and Habra M, et al (2012) Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas J Clin Endocrinol Metab 97 4040–4050 https://doi.org/10.1210/jc.2012-2356 PMID: 22965939 PMCID: 3683800
16. O’Kane G, Ezzat S, and Joshua A, et al (2019) A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: the SNIPP trial Br J Cancer 120 1113–1119 https://doi.org/10.1038/s41416-019-0474-x PMCID: 6738062
17. Baudin E, Goichot B, and Berruti A, et al (2021) First international randomized study in malignant progressive pheochromocytoma and paragangliomas (FIRSTMAPPP): an academic double-blind trial investigating sunitinib Ann Oncol 32(suppl_5) S621–S625 https://doi.org/10.1016/j.annonc.2021.08.702
18. Van Hulsteijn L, Niemeijer N, and Dekkers O, et al (2014) (131)I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: systematic review and metaanalysis Clin Endocrinol (Oxf) 80 487–501 https://doi.org/10.1111/cen.12341
19. Agrawal A, Rangarajan V, and Shah S, et al (2018) MIBG (metaiodobenzylguanidine) theranostics in pediatric and adult malignancies Br J Radiol 91(1091) 20180103 https://doi.org/10.1259/bjr.20180103 PMID: 30048149 PMCID: 6475939
20. Satapathy S, Mittal B, and Bhansali A (2019) Peptide receptor radionuclide therapy in the management of advanced pheochromocytoma and paraganglioma: a systematic review and meta-analysis Clin Endocrinol (Oxf) 91(6) 718–727 https://doi.org/10.1111/cen.14106 PMID: 31569282
21. Ferrara A, Lombardi G, and Pambuku A, et al (2018) Temozolomide treatment of a malignant pheochromocytoma and an unresectable MAX-related paraganglioma Anticancer Drugs 29(1) 102–105 https://doi.org/10.1097/CAD.0000000000000570
22. Tena, I, Gupta G, and Tajahuerce M, et al (2018) Successful second-line metronomic temozolomide in metastatic paraganglioma: case reports and review of the literature Clin Med Insights Oncol 12 1179554918763367 https://doi.org/10.1177/1179554918763367 PMID: 29720885 PMCID: 5922490
23. Clarke M, Weyant R, and Watson C, et al (1998) Prognostic markers in pheochromocytoma Hum Pathol 29(5) 522–526 https://doi.org/10.1016/S0046-8177(98)90070-3 PMID: 9596278
24. Ohji H, Sasagawa I, and Iciyanagi O, et al (2001) Tumour angiogenesis and Ki-67 expression in phaeochromocytoma B J U Int 87(4) 381–385 https://doi.org/10.1046/j.1464-410x.2001.00102.x
25. Salmenkivi K, Heikkilä P, and Haglund C, et al (2003) Lack of histologically suspicious features, proliferative activity, and p53 expression suggests benign diagnosis in phaeochromocytomas Histopathology 43(1) 62–71 https://doi.org/10.1046/j.1365-2559.2003.01645.x PMID: 12823714
26. August C, August K, and Schroeder S, et al (2004) CGH and CD 44/MIB-1 immunohistochemistry are helpful to distinguish metastasized from nonmetastasized sporadic pheochromocytomas Mod Pathol 17(9) 1119–1128 https://doi.org/10.1038/modpathol.3800160 PMID: 15167935
27. Tavangar S, Shojaee A, and Moradi Tabriz H, et al (2010) Immunohistochemical expression of Ki67, c-erbB-2, and c-kit antigens in benign and malignant phaeochromocytoma Pathol Res Pract 206 305–309 https://doi.org/10.1016/j.prp.2010.01.007 PMID: 20189725
28. Cantalamessa A, Caobelli F, and Paghera B, et al (2011) Role of (18)F-FDG PET/CT, (123)I-MIBG SPECT, and CT in restaging patients affected by malignant pheochromocytoma Nucl Med Mol Imaging 45(2) 125–131 https://doi.org/10.1007/s13139-011-0083-y PMID: 24899991 PMCID: 4043021
29. Shulkin B, Thompson N, and Shapiro B, et al (1999) Pheochromocytomas: imaging with 2-[Fluorine-18]fluoro-2-deoxy-D-glucose PET Radiology 212 35–41 https://doi.org/10.1148/radiology.212.1.r99jl3035 PMID: 10405717
30. Ezuddin S and Fragkaki C (2005) MIBG and FDG PET findings in a patient with malignant pheochromocytoma: a significant discrepancy Clin Nucl Med 30 579–581 https://doi.org/10.1097/01.rlu.0000170060.52675.14 PMID: 16024963
31. Hescot S, Curras-Freixes M, and Deutschbein T, et al (2019) Prognosis of malignant pheochromocytoma and paraganglioma (MAPP-Prono Study): a European network for the study of adrenal tumors retrospective study J Clin Endocrinol Metab 104(6) 2367–2374 https://doi.org/10.1210/jc.2018-01968 PMID: 30715419





