Cancer and the metastatic substrate
Francisco Arvelo 1,2, Felipe Sojo 1 and Carlos Cotte2
1Life Sciences Centre, Institute for Advanced Studies Foundation [Fundación Instituto de Estudios Avanzado]-IDEA, Apartado 17606, Caracas 1015-A, Venezuela
2Tumour Biology Culture and Tissue Laboratory, Experimental Biology Institute, Central University of Venezuela, Apartado Apartado 47114, Caracas 1041-A, Venezuela
Correspondence to: Francisco Arvelo. E-mail: franarvelo@yahoo.com
Abstract
Seventy percent of cancer patients have detectable metastases when they receive a diagnosis and 90% of cancer deaths result from metastases. These two facts emphasise the urgency for research to study the mechanisms and processes that enable metastasis. We need to develop a greater understanding of the cellular and molecular mechanisms that cause metastasis and also we need to do more. We must also consider the micro- and macro-environmental factors that influence this disease. Studying this environmental context has led us to update the ‘seed and soil’ hypothesis which dates back to the 19th century. This theory describes cancerous cells as seeds and the substrate as the soil in target organs though this may seem antiquated. Nonetheless, the tissue specificity that researchers have recently observed in metastatic colonisation supports the validity of the seed and soil theory. We now know that the metastatic potential of a tumour cell depends on multiple, reciprocal interactions between the primary tumour and distant sites. These interactions determine tumour progression. Studies of metastasis have allowed us to develop treatments that focus on therapeutic effectiveness. These new treatments account for the frequent metastasis of some tumours to target organs such as bones, lungs, brain, and liver. The purpose of this review is first to describe interactions between the cellular and molecular entities and the target organ tumour environment that enables metastasis. A second aim is to describe the complex mechanisms that mediate these interactions.
Keywords: cancer, metastasis, tumour micro-environment, tumour progression, tropism, exosomes, micrometastasis
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (
Published: 08/12/2016; Received: 20/05/2016
Introduction
The genetic paradigm of cancer posits that tumours result from multiple mutations in a single normal cell. These mutations alter the genotype of the cell and transform it into a malignant phenotype [1]. The time required for this process to develop varies greatly, but it begins with the cloning of cells. Over months, years, or decades, this cloning results in the formation of a primary tumour [2]. We now recognise that malignant mutation, development, and transformation only occurs in progenitor cells called stem cells [3]. Mutations though in somatic cells do not produce cancer as their short half-life illustrates. Cells differentiate, mature, carry out their functions, and complete their life cycles when they die in apoptosis. The aggressiveness and metastatic power of a tumour depends on the maturity level of the stem cell that produced the mutation. Tumours derived from stem cells in early maturity will have a more heterogeneous phenotype and will metastasise quickly. Tumours derived from a more mature stem cell will have a more homogeneous phenotype and are less metastatic [4]. The biological heterogeneity of cellular populations that comprise malignant neoplasms varies widely. The notable properties of these cellular populations include their cellular surface, antigenicity, immunogenicity, proliferative index, and their sensitivity to antitumour agents. Also significant is phenotypic expression, which in combination with the aforementioned factors, allows tumours to invade other tissues.
The metastatic cascade begins in the primary tumour via local invasion characterised by several factors including the mechanical pressure exercised by proliferating tumour tissue. The action of proteolytic enzymes reduces the molecular organisation of barriers and lowers resistance to invasion. The capacity of metastatic cells to displace other cells is also a factor in the metastatic cascade [5]. This dynamic invasion process produces a Darwinian evolutionary selection in which cells acquire changes to their genetic material. These changes confer an advantage which over time becomes more common in the tumour through selection. Genetic instability thus characterises these cells and allows them to develop the capacity to invade and metastasise [6]. Metastasis develop and evolve as tumour cells spread and establish themselves in distant organs. Metastasis not only determine the prognosis and life expectancy of patients, but they also dictate the clinical outcomes of most tumours [7, 8]. Current research that examines cellular and molecular processes is critical, but we must also study the cellular, tissue, and organ environments. All of these research areas are essential to understanding cancer and finding better and more effective treatments.
I. Metastatic dissemination
The spread of tumour cells: Malignant tumours spread through the circulatory and lymphatic systems via the intravasation of tumour cells. Angiogenesis facilitates this process, and it results from the development of microcirculation in neo-formed vessels that form a fenestrated endothelium. These vessels are unstable intercellular unions, and form a discontinuous basal membrane that is sometimes absent [9]. Tumour progression requires that invasion coincide with an increase in vascularisation that provides nutrients and factors essential to the growth of tumour cells. Neovascularisation occurs early in tumour progression and is detectable when diseases such as ‘in situ’ carcinomas are incipient [10]. Tumour cells stimulate endothelial cell proliferation and vessel formation through various factors. Among the best known of these factors are vascular endothelial growth factor, or VEGF, IL-8, and TNF-α. Endothelial cells also produce other growth factors, such as FGF, which also promotes the growth of tumour cells [11].
Once tumour cells penetrate vessels, they can reach distant organs and proliferate. Metastasis is a highly inefficient process; even when millions of cells detach and migrate from a tumour, only a small fraction will survive and form a new tumour. This paradox is common in nature as all species use a large number of organs or specialised parts to survive. In cancer, great numbers of cells migrate from the primary tumour ensuring that some have a chance to survive and form new tumours. Although inefficient, cell migration from primary tumours is so effective that metastasis is the main cause of cancer deaths. Tumour cells that migrate may die from one of four fundamental causes. Firstly, cells are usually linked with other cells or with the surrounding environment while migrating cells are not. Consequently, migrating cells may die in a type of apoptosis that affects detached cells, anoikis. Tumour cells may also die because they are larger than blood cells or they may undergo cytoskeletal changes that cause cell death. Finally, migrating tumour cells are also vulnerable to immunological mechanisms [12]. The importance of epigenetic mechanisms in tumour progression is also becoming more evident, especially in the context of the cellular and extracellular environments [9]. Angiogenesis is one of the distinct and crucial events in tumour progression and cancer malignancy [13].
Adhesiveness between normal and tumour cells: Research has examined specifically how tumour cells adhere to the microvascular endothelium of the target organ. Specific molecules on the surfaces of both types of cells are responsible for adhesion and determine the specific site of metastasis. Three studies identified specific characteristics of circulating tumour cells, endothelia, target organs, and the microenvironment of each organ. Data from these studies indicate that these characteristics play a role in determining the sites of secondary tumour foci [14, 15, 16]. The cells capable of forming metastasis are extravasated tumour cells in close contact with blood vessels. Soluble factors secreted by tumours induce the formation of tumour foci via endothelial activation. This process is mediated by FAK (which upregulates E-selectin) and promotes the adhesion of tumour cells to the endothelium [17]. Moreover, endothelial activation by IL-1α, IL-1β, or TNF-α induces expression of E-selectin and P-selectin, as well as VCAM-1 and ICAM-1 on the surface of endothelial cells. The binding of these molecules to their ligands on tumour cells can promote contact and adhesion of tumour cells to endothelial cells [18]. Malignant cells targeting the lungs express high levels of Angpt14 and VEGF-A factors, which hinder cell-endothelial cell unions and facilitate extravasation [19]. EREG, COX2, MMP1, and MMP2 molecules also promote extravasation and metastasis [20]. Extravasation can also result from the interaction of tumour cells and platelets, in which TGF-ß activates TGF/Smad and NF-kB in cancerous cells. This process induces the epithelial-mesenchymal transition in tumour cells, stimulating extravasation [21]. Monocytes/macrophages recruited by tumour cells promote the establishment of metastatic breast cancer tumour cells in the lungs [22].
The F4/80 , CD11b , and Gr1 macrophages are associated with metastasis and secrete VEGF-A. This process promotes extravasation and the establishment and growth of tumour cells, possibly by increasing endothelial permeability [23]. However, soluble factors secreted by primary tumours induce recruitment of bone marrow-derived cells (BMDCs) which include immature myeloid cells, neutrophils, and monocytes. This process occurs in distant organs and results in the formation of a premetastatic niche, which ensures the survival and growth of tumour cells [24, 25, 26].
Non-random metastatic distribution patterns: Researchers have proposed two theories to explain the metastatic selectivity of tumour cells:
I. The mechanistic theory: The relative haemodynamic position of each organ in relation to the primary tumour site determines metastatic selectivity. This theory posits that tumour cells leave the primary tumour by following the circulatory and/or lymphatic drainage routes. Tumour cells then stop at the first organ they encounter, in a process that is non-specific. Consequently, the first organ along the drainage route will be the site of the greatest number of metastases. Anatomical and mechanical factors support this theory and are significant in determining the metastatic patterns of multiple types of tumours. For example, gastrointestinal tumours start as a localised tumour in the digestive tract mucosa and grow. These tumours then invade the deepest layers of the digestive tract mucosa until reaching the serosa. When the tumour crosses the intestinal wall, it can then invade any organ within or outside the abdomen.
II. The lymphatic dissemination theory: Lymphatic dissemination occurs when tumour cells reach the network of lymphatic vessels that surround the colon, allowing lymph drainage to multiple lymph node regions. Dissemination via the lymphatic vessel network occurs in a sequence affecting the closest lymph nodes first before spreading to more distant ones.
III. Haematogenous dissemination occurs via tumour cells which pass into the bloodstream. Generally the first area they spread to is the liver followed by the lungs and bones because of the fact that the venous return of the intestinal tract is produced though the portal system. However, tumours that originate in the distal part of the rectum can give rise to an initial metastasis in the lungs because the inferior rectal vein drains into the inferior vena cava and not into the portal venous system [27].
Organ tropism.- In 1889, Stephen Paget [28] postulated his hypothesis classically known as ‘seed and soil’, asserting that the capacity of metastatic colonisation is determined by the inherent properties of tumour cells, comparing cells to ‘seeds’ and the place where they grow as the ‘soil’ which is the tissue substrate. Paget examined 735 women who had died from breast cancer and concluded that the distribution of metastases is not because of the chance that the cells were not distributed randomly, but rather because of special select microenvironment, the tissue substrate. He concluded that the environment played an important role in the formation and growth of the metastasis. This was backed up by experiments with animal models by injecting them with tumour cells with an affinity for a certain organ, hence, proving that the colonisation was organ specific [29, 30]. This tropism was proven experimentally with the pioneering work of Fidler and Nicolson (1976) who used two cellular variants of the murine melanoma B16: one with low growth potential, B16-F1; and the other with high growth potential, B16-F10. These cells were injected in the spleens and hearts of mice, proving that the B16-F10 cells exclusively form metastasis in the lungs, while the B16-F1 cells formed extrapulmonary metastases. These results determined that the cells did not accumulate in the first organ they found on their journey but rather the cells specifically accumulated in the lungs [31].
Premetastatic niche preparation.- Paget’s hypothesis gave rise to the idea that before the metastatic dissemination occurs, the cells of the primary tumour liberate cell mediators, nowadays called exosomes. These can travel in the circulatory system and encourage the formation of the premetastatic niche for tumour growth [32, 33]. Unlike soluble factors secreted by the cells, the exosomes contain groups of functional molecules that serve as intercellular communicators not only at a local level but also on a systemic one, apart from serving as protection for the transported molecules. The majority of the prokaryotic and eukaryotic cells liberate exosomes, including colorectal [34], lung, breast, glioblastoma, ovary, and melanoma cancer cells [35]. The exosomes are membrane vesicles that originate within the cells in endosomal compartments called multivesicular bodies [36]. Likewise, the exosomes transfer bioactive molecules to both distant and proximal receptor cells thereby altering cell’s signalling and the phenotype. Fibroblast growth factor (FGF), hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF) [37, 38], and epidermal growth factor (EGF) [39] are what stand out most along with the mutated EGFR receptors [40, 41] and HGFR [42]. It has been proven that the exosomes deriving from both normal cells and cancer cells are mediators of the metastatic process as they can promote angiogenesis [43, 44], the invasion [45, 46], and proliferation of receptor cells to support the growth of the tumour [47, 48]. It has also been suggested that exosomes that derive from cancer because of their pleiotropic character could be involved in the development and progression of the tumour through the processes that enable the following: (a) tumour cells escaping from the immune system aid in launching the inflammatory response; (b) act on the differentiation of the fibroblasts and the mesenchymal cells; (c) improve the metastatic evolution of the tumour via the promotion of the epithelial/mesenchymal transition of the tumour cells and prepare the niches of the tumour cells for their new anatomical locations [49, 50].
The exosomes of a highly metastatic melanoma increased the metastatic capacity of the primary tumour because of the influence of the progenitor cells of bone marrow via the MET receptor tyrosine kinase. These exosomes induced a vascular phenotype in the premetastatic site and reprogrammed the progenitor cells of the bone marrow that expressed c-kit, the receptor tyrosine kinase Tie2, and MET. The reduction of the expression of MET in the exosomes reduced the pro-metastatic capacity of the progenitor cells of the bone marrow. On the other hand, the progenitor cells of the bone marrow of people with metastatic melanoma with phenotype CD45 (-) C-KIT (Low/ ) TIE2 ( ) presented an elevated expression of MET. Likewise, the regulators of membrane transport RAB1A, RAB5B, RAB7, RAB27A (RAS-related GTP-binding proteins) and the formation of exosomes were highly expressed by the melanoma cells [41]. The integrins contained in the exosomes that participate in the formation of the premetastatic niche could be used to predict organ-specific metastasis. Exosomes of the lungs, livers, and brains of mice and humans fused with the resident cells such as the epithelial cells and lung fibroblasts, Kupffer cells and endothelial brain cells. The exosomes with integrins α6β4 and α6β1 were associated with lung metastasis, while the integrin αvβ5 was associated with liver metastasis. Likewise, it was proven that the recruitment of integrins by resident cells activates the phosphorylation of Src and the expression of the proinflammatory gene S100 (51). The human renal cancer stem cells with a phenotype marker CD105 gave rise to microvesicules that provided an angiogenic phenotype activated by the endothelial cells and promoted the formation of the premetastatic niche. These renal carcinoma cells injected into mice with severe combined immunodeficiency gave rise to the formation of lung metastasis [52]. A model of a pancreatic adenocarcinoma BSp73ASML in a rat was used to analyse the role of the CD44v variant isoform in the formation of a premetastatic niche. The CD44v is a requisite for the formation of a water-soluble matrix which in collaboration with the exosomes promotes the accumulation of leukocytes, the activation of endothelial and stroma cells, with the exosomes being the principal elements in the preparation of the premetastatic niche. The soluble fraction of the tumour serves as an exosome carrier which at the same time are carriers of chymosins and proteases necessary for the preparation of the premetastatic niche. The CD44v variant isoform could be used as a marker in tumour progression [53]. The pancreatic ductal adenocarcinoma is highly metastatic and of poor prognosis, and its exosomes induced the formation of the premetastatic niche in the liver with the subsequent formation of metastases in mice. The recruitment of exosomes derived from this tumour by the Kuffer cells induced the secretion of the transforming growth factor β and an overregulation of the production of fibronectin by the Kuffer liver cells. Initially, this gave rise to a fibrotic microenvironment and an increase in the recruitment of macrophages from the bone marrow. The macrophage migration inhibitory factor (MIF) was highly expressed by the exosomes of the pancreatic tumour and its blockage prevents the premetastatic niche formation and subsequently metastasis. In patients whose pancreatic tumours did not progress, MIF was greater in the exosomes of the patients with early stage pancreatic tumours that developed metastasis in the liver. MIF can be used as a prognostic marker in the development of liver metastasis in patients with pancreatic cancer [54].
II. Metastasis in target organs
Those organs which are frequent targets for metastases will be reviewed below. We will also give consideration to each of their particular and specific treatments. This will reaffirm the complexity of a illness that it is not lineal but contextual.
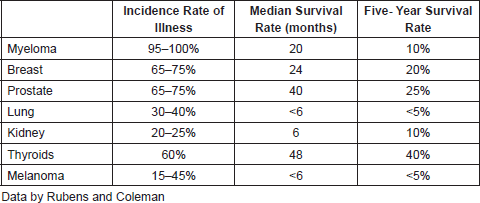
Metastasis in the bone.- The bones are the parts of the organism that are most affected by cancer where metastases occur from malignant breast, prostate, lung, melanoma, myeloma, kidney, and thyroid tumour types. Table 1 shows the incidence of bone metastases as a function of the primary tumor.
The predisposition of some cancers to produce metastasis in the bone, specifically in the medullary compartment, is because of the capillary structure of the bone marrow. There is slow blood flow facilitating the cells to be retained in the vascular sinusoids which are very spacious [55]. Bone marrow is a great source of growth factors such as TGF-β, IGF, of fibroblasts FGF, those derived from platelets (PDGF), morphogenic bone protein (BMPs), plus calcium which provides an appropriate environment for the growth of tumour cells [56]. A new model has been proposed to explain the preference of tumour cells for producing metastases in certain organs, affirming that not only is the bone microenvironment important, but also that the actual primary tumour produces exosomes and certain factors that prepare the target organ with a premetastatic niche in order to be able to host the disseminated tumour cells [57]. As a response to the soluble factors liberated by the haematopoietic precursor cells and the macrophages, present in the bone microenvironment, an adequate environment is created i.e. a ‘fertile soil’ favourable for invasion by metastatic cells [58]. The tumour cells that produce bone metastasis express the chemokine receptor CXCR4, motif chemokine receptor 4 CXC in its membrane which responds to the chemoattractant signals generated by its ligand SDF-1, also known as CXCL12. This factor is secreted both in the bone microenvironment by the osteoblasts, fibroblasts, haematopoietic stem cells, endothelial cells in the bone marrow, similar to the higher levels in the premetastatic niche. In turn the ligand SDF-1 also attracts the haematopoietic progenitor cells of the bone marrow and the endothelial progenitor cells to the metastatic site [59]. In addition, the integrin expression αvβ3 in the metastatic cells of prostate carcinoma facilitates the union to bone matrix proteins–collagen, fibronectin, vitronectin and osteopontin or OPN-, permitting the tumour cells to grow inside the bone. Not only are many factors of the bone microenvironment involved in this, but also factors secreted by the actual tumour cells [60].
With respect to the primary tumour, we see different factors are produced that stimulate the activity of osteoblasts or osteoclasts thereby giving rise to the formation of osteolytic and osteoblastic bone metastasis. Mixed metastases can also develop from both components. For different types of cancer, the lesions seen can be very different [61]. The metastatic cells of osteolytic types are characterised for production of an increased activity of the osteoclasts in the host giving rise to the destruction of the bone. Histologically, this is characterised by the presence of the osteoclasts eroding the bone in the tumour-bone interface [62]. Breast, lung, and kidney cancer preferably trigger the activation of the osteoclasts resulting in the appearance of osteolytic lesions [63]. The protein related to the parathyroid hormone (PTHrP), liberated by the metastatic cells, is the principal mediator of osteoclast activation in osteolytic metastasis. This protein stimulates the osteoblasts so that they express the ligand for the receptor activator of nuclear factor -kB or RANK more and the osteoprotegerin OPG less. The result is the activation of osteoclastogenesis and the increase in bone resorption. The destruction of the bone provokes the liberation of growth factors and the increase in the concentration of calcium. All these factors unite to their receptors in the membrane of the tumour cells stimulating the growth and the synthesis of PTHrP, such as multiple cytokines -like IL-1, IL-6, IL-11, IL-8-, which induce osteolysis to which the vascular endothelial growth factor or VEGF is added. This creates a viscous circle that induces the growth of tumour cells and the activity of the osteoclasts [64, 61, 62]. Bone metastasis from breast cancer affects approximately 85% of the patients with this illness. It has been found that hypoxia is associated with bone metastasis in patients with oestrogen-negative breast cancer. A high expression of lysyl oxidase LOX is associated with bone metastasis from an osteolytic lesion. LOX has been identified as a regulator of the osteoclastogenesis independent of RANK that leads to the formation of premetastatic focal lesions, which provide the platform for the tumour cells to colonise and establish as metastasis [65].
Table 1. Incidence and prognosis of bone metastases of different types of cancer.
In the case of osteoblastic metastasis, this is a hallmark of prostate cancer, whose malignant cells are characterised by producing an increase in osteoblastic activity which further leads to an increase in ectopic bone formation. In this kind of metastasis the presence of a large number of osteoblasts near the tumour cells can be observed [66]. The main modulator of these metastases is the protein Endotelia-1 or Et-1, which is liberated by the metastatic cells and stimulates the proliferation of the osteoblasts through an inhibitor of the Wnt cellular signalling pathway in charge of the differentiation and activation of the osteoblasts. This inhibition is called dickkopfs or DKK1 [67]. The tumour cells also produce other osteoclast-activating factors such as BMPs and PDGF. Once they are differentiated, the osteoblasts start forming a new non-mineralised matrix on the already existing bone which contains growth factors and other proteins that attract tumour cells and enable them to survive and proliferate. The TGF-β1 is another factor that participates in the progression and development of prostate carcinoma metastasis, facilitating osteoblastic bone metastasis, and which has been proven experimentally [68]. The anti-tumour efficiency of a specific inhibitor of the kinase receptor-1 of TGF-β1, which is capable of controlling the growth of the prostate carcinoma cells in the bone [69], has also been proven. The urokinase uPA participates in the osteoblastic metastasis by having the capacity to separate and activate TGF-β which enables both the differentiation of osteoblasts and osteoclasts and also the growth of malignant cells [70]. The prostate specific antigen PSA also separates and activates TGF-β and PTHrP in the extreme N-terminus. This way, not only is the re-absorption mediated by PTHrP prevented, but it converts it into a stimulating factor for bone deposition [71]. Likewise, the PSA can separate IGFBP-3, the IGF-1 binding protein which cause an increase in the free IGF-1 levels, thus increasing osteoblastic activity [72].
In the treatment of prostate carcinoma, there is a close relationship with androgens which is why their neutralisation or deprivation through chemical or surgical means has become the treatment for advanced stages of the illness. The initial response to the treatment has been satisfactory as it has produced a significant clinical regression in the illness [73]. Nevertheless, despite a good response to the treatment, a relapse usually occurs after castration [74] which makes the illness progress to a phase that is insensitive to the neutralisation of hormones called castration-resistant prostate cancer CRPC. It determines the prognosis which can be dire with a survival rate of 16 to 18 months. The other option for these patients is chemotherapy which will be applied depending on which cells the treatment is aimed at. These are divided into three categories: 1) epithelial cells, whose treatment is based on the use of cytotoxic agents such as docetaxel and cabazitaxel [75, 76]; 2) stroma cells which include endothelial, osteoblastic, and osteoclastic cells. The cytotoxics used are thalidomide which is an anti-angiogenic [77]; bevacizuman, a monoclonal antibody against VEGF-A [78]; antrasentan, an osteoblast proliferation inhibitor [79]; denosumab, a monoclonal antibody against RANKL; the activating receptor ligand for the nuclear factor ϰB [80]; zoledronic acid, belongs to the group of bisphonates which inhibit the action of the osteoclasts in the prostate carcinoma cells and encourage an increase in bone density [81, 82]; 3) block the androgen receptor activation or AR which is expressed both in the prostate carcinoma cells and the microenvironment cells. For this purpose abiraterone is used which suppresses testosterone production [83], and MDV3100 or enzalutamide, an androgen receptor antagonist or AR, which blocks signalling as it inhibits its translocation to the nucleus and prevents binding to the DNA [84].
Metastasis in the lung.-The state of the organs has a leading role in the development of a metastatic lesion as tumour cells are more capable of developing growth points if they have a conducive microenvironment. This specification is determined by local growth factors from hormones or cytokines secreted by target organs, by adhesive interactions of the endothelium with the tumour cells, or by susceptibility in the tissue that facilitates the adhesion of the tumour cells [85]. Most likely there is a combination of the anatomical role and the specific tissue for a tendency for metastasis in the lungs [86]. The tumours that originate in the breasts, bladder, colon, kidneys, head and neck, and melanomas have a tendency to cause metastasis in the lung. According to the theory of Paget, the lung is a good substrate for developing metastasis because of it being a highly vascularised tissue, well oxygenated and nurtured, as well as being a tissue rich in alveolar macrophages [87]. The myeloid cells BMDC are the first defence barrier against respiratory pathogens which act against infections and secrete pro-inflammatory cytokines such as interleukin-6 and tumour necrosis factor-alpha or TNF-α. Both can increase permeability and tumour angiogenesis and their expression can be stimulated remotely by tumours [88]. The myeloid cells can also express VEGFR1 in a similar way to how the endothelium cells and some cancerous cells do in addition to secreting proteolytic enzymes such as MMP-9 in response to the activation of VEGFR1 through its ligands. It has been proven in knockout mice that MMP-9 and the TNF-α are crucial for metastasis in the lung as in its presence, cancerous cells can become established because of abundant pulmonary vascularisation and grow as tumour nodules [88]. Hiratsuka et al proposed the concept that previously preparing the lung tissue happens before metastasis actually occurs. In an experimental model to study metastases, it has been proven that the activation of the endothelium cells and lung macrophages, caused by the cells from the primary Lewis tumour or LLC (Lewis lung carcinoma), encourage the formation of metastasis in the lungs. Additionally, the suppression by VEGFR1 or MMP-9 reduces the metastasis in the lung [89]. Likewise, in the Lewis model of lung cancer, areas of the accumulation of clusters of VEGFR1, BMDC have been described along with fibronectin causing a pre-metastatic niche. These areas preferably harbour LLC cells, and it is observed that the VEGFR1 block prevented the formation of the metastasis [24, 90, 91]. Members of the VEGF family and the placental growth factor PlGF have been characterised as angiogenesis modulators in many tumours [92]. These growth factors bind to the VEGF receptors in the endothelial cells encouraging proliferation, survival, and migration. VEGF binds to VEGFR1, VEGFR2 and PNR (neuropilins), while PlGF only binds to VEGFR1, NRP-1, and NRP-2 [93]. The expression of VEGFR1 can be constitutive or induced by the expression of VEGF and PIGF caused by hypoxia which is accompanied by tumour growth [94]. On the other hand, the lysyl-oxidase, induced by hypoxia to the recruitment of the BMDC to the lungs, is a phenomenon that occurs during the formation of the metastasis. This supports the idea that the formation of the metastasis in the lung depends on the respiratory tract which include the accumulation or activation of the BMDC cells in the lung tissue [95]. By using a melanoma in an experimental model an increase in metastasis could be observed because of a high expression of the vascular cell adhesion molecule VCAM-1 in the microvasculature of the lung. An increase in the frequency of pulmonary metastasis has been observed when the animals have been inoculated with tumour cells that were previously treated with pro-inflammatory cytokines [96].
The treatment of patients with pulmonary metastasis is based on three modalities: a) prophylactic radiotherapy used in low doses on tumours in the lungs, as well as in pre-surgical pulmonary lesions; b) the resection of pulmonary metastasis through surgery which is only done when there are no extra-pulmonary metastases. It is an appropriate treatment for primary malignancy and if there is tolerance on the patient’s part, a complete recession of all the pulmonary metastases is carried out; c) chemotherapy is the standard treatment for multiple pulmonary metastases, especially in tumours with a high rate of proliferation [97]. In a murine model of breast tumour, the combined use of cyclophosphamide or CP and dabigatran etexilate, a direct inhibitor of thrombin, showed a significant synergistic effect. The tumours were significantly smaller and produced less pulmonary metastases when compared to mice only treated with one of the drugs [98]. Patients with triple-negative breast cancer for oesterogen receptor (ER)-, progesterone receptor (PR),- and HER2- have a poor prognosis as they frequently develop metastasis. The cells from this cancer, treated with selumetinib, a MEK inhibitor, showed a growth inhibition, and it was determined that significantly fewer pulmonary metastases formed in the mice than in the control mice. This MEK inhibitor therefore represents a potential alternative for the prevention of metastasis [99]. Eribulina, a microtubule polymerisation inhibitor, was used to measure the metastatic activity of the cells of a breast cancer ER-, PR, HER2- in an experimental model ‘in vivo’ with pulmonary metastasis. The pre-treatment ‘in vitro’ with this inhibitor caused an inversion of EMT and an induction of MET with the following reduction in the number of pulmonary metastases [100]. The ‘micro-ARN’ are conserved ARN that regulate different cellular processes whose dysfunction is involved in the development and progression of breast cancer. The chemokine receptor CXCR7 is involved in various biological processes such as cell survival, adhesions and mobility, whose over-expression has been observed in different cancers, like breast, lung, and prostate cancer. The results obtained both ‘in vitro’ and in a pulmonary metastasis model ‘in vivo’ confirmed that miR-101 is an inhibitor of growth and the invasion of the breast cancer cells through the inhibition of the CXCR7-STAT3 pathway. These results show a potential role of miR-101 as a therapeutic alternative for breast cancer [101].
Metastasis in the brain.- This type of metastasis is very common, making up 80% of those produced by primary lung tumours, breast cancers, and melanomas [102]. Because of the high incidence of asymptomatic brain metastasis, it is difficult to calculate the true prevalence, but different studies estimate that between 15 and 25% of cancer patients develop metastasis in the brain [103, 104]. The survival rate of patients with untreated brain metastasis is two months but could be extended to six months with conventional radiotherapy and chemotherapy treatment [105]. Numerous studies have highlighted the mechanisms that actively participate in the recruitment of tumour cells in the brain, having also established that the development of metastasis in the brain is not random. It is fundamentally a coordinated accumulation of mutations that enable the cancer cells to become established within the central nervous system. The carcinomas that produce metastasis in the brain have a preference for certain regions in the brain: A 80% in the cerebral hemispheres, 15% in the cerebellum, and 5% in the brain stem [106]. In the case of breast cancer, the appearance of brain metastasis usually occurs after the primary tumour has been surgically removed and after a latency period of two or three years has passed [107]. Additionally, there are different characteristics between breast cancer brain metastasis and the site of the primary tumour, expressing an increase in Ki67, a greater micro-vascular density, and the expression of micro-ARN [108, 109]. In HER-2 breast tumours the possibility of developing brain metastasis is increased as it possibly has an affinity for neural tissue, and this occurs on average in 20% of breast cancers [110]. The difference between the primary tumour of breast carcinoma, negative for HER-2 but positive for brain metastasis, has been reported in 24% of cases which show lower survival rates [111]. The increase in the brain metastasis rate in breast cancer HER2 highlights the tropism of the HER2-positive cells to the brain parenchyma [112, 113].
In experiments with cell lines, the overexpression of HER-2 increases the production of TGF-β, leading to the activation of TGF/Smad and the expression of E-cadherin, including snail, slug, and ZEB-1. The inhibition of HER-2 by curcurbitacin 2 leads to the inhibition of brain metastasis ‘in vivo’ [114]. On the other hand, HER-2 contributes to the epithelium/mesenchyma transition EMT through the production of TGFβ, which is its regulator [115]. The blood-brain-barrier, known by its initials ‘bbb’, is an obstacle for the infiltration of tumour cells because of the endothelial tight junctions. In studies using an animal model and an MRI scan, it has been shown that approximately 1.5% of the injected malignant cells formed metastasis in the brain. Among 95% of them were located throughout the brain blood vessels whilst the colonies that became established did so in the brain parenchyma. This result suggests that the basal vascular membrane represents a fertile ‘soil’ for the formation of metastasis [116, 117, 118]. The extravasation of tumour cells is facilitated by the expression of COX-2 by part of the endothelial cells which in turn induce the expression of metalloproteins by the tumour cells [119]. The overexpression of αB-crystallin by the breast cancer metastatic cells induced an increase in adhesiveness on the endothelial cells of the microvasculature of the brain [120].
The junctional adhesion molecule A or JAM-A, is a transmembrane protein that belongs to the super family of immunoglobulins, Ig, which is dysregulated in the breast cancer metastatic cells of the brain. Its expression is associated with the patients’ evolution and as a prognostic indicator [121]. The alpha 2 6-sialiltranferasase ST6GALNAC5 media, specifically in the brain metastasis and their expression in breast cancer cells, increases their adhesiveness to endothelial cells in the brain as they pass through the blood-brain barrier bbb. The participation of this sialyltransferase in the brain highlights the role of the glycation in the cell surfaces of the interactions that occur in organ-specific metastasis [122]. On the other hand, the role of astrocytes in breast tumour cell survival in the brain must be highlighted. Once they have passed through the bbb they are surrounded and localised by reactive astrocytes through the overexpression of the metalloproteins or MMP, such as MMP-2 and MMP-9 [123]. In addition, the astrocytes encourage tumour growth by the secretion of cytokines, la heparanasa, neurotrophic factors, TNFα, TGFβ, IL6, etc [124]. The reactive astrocytes defend themselves against the metastatic invasion from the overexpression of the plasmin as its anticoagulant effect stimulates the paracrine secretion of the cell death factor FasL and of the inactivation of L1CAM secreted by the tumour cells. This prevents the establishment of tumour cells throughout the blood vessels. However, the tumour cells survive in the brain parenchyma protected from the plasmin by the expression in the tumour cells of the serpins, a serine protease [125]. Likewise, the astrocytes can promote the survival and growth of the metastatic tumour cells from the neurotransmitter γ-aminobutyric or GABA. The GABA receptors present in the breast cancer cells connect to them and then catabolise to confer a metabolic advantage for the growth of metastatic cells [126]. It is proposed that the brain parenchyma provides some kind of nourishing or growth factor that vitalises the metastatic tumour [127]. The altered metabolism is characteristic of the cancer cells. It develops a strategy which enables the metastatic brain cells to survive and proliferate. The metastatic cells do this through a process of anaerobic glycolysis, whilst normal cells do this because of the aerobic oxidation of the pyruvate [128]. Moreover, the aggressiveness of the triple negative cancer tumours (TN) are associated with a high rate of metastases to the central nervous system [129]. It has been suggested that the androgens activate the brain parenchyma astrocytes in order to facilitate the establishment of the breast cancer TN cells. This would explain why young patients with TN that present high levels of oestrogen have a higher risk of suffering brain metastasis. Possibly, the development of these metastasis could depend more on the hormonal profile of the patient than the intrinsic factors of the tumour cell [130].
The resistance to systemic therapy by metastatic brain tumours has been attributed to the bbb barrier. It was supposed that this was intact in the blood vessels of the metastatic lesion which prevented the distribution of the drugs. Nevertheless, it is now known that the presence of an intracranial metastasis modifies the vascular integrity and that the bbb then does not show normal characteristics, consequently it is known as the blood-tumour barrier ‘btb’ [131]. This barrier presents an increase in the permeability, reduction of blood flow, and an increased expression or reduced affluence of efflux transporters [132, 133]. The structure of the vascular network within the brain tumours is anatomically and functionally different. Anatomically the tumours promote the formation of new deformed blood vessels that lack the classical structure of the bbb. The new deformed blood vessels do not have an adequate contact with the astrocytes and in addition present fenestration or pores that enable the free movement of molecules to the brain [134, 135]. In functional terms, the vascular network within the brain tumour reduces the expression of proteins of strong bonds such as ZO-1. These bonds serve as an anchor for the union of occludin with the endothelial membrane consequently increasing the vascular permeability [136, 137]. In adults brain metastasis clinically make up more than half of all brain tumours. The present treatment options include surgical removal, radiotherapy, and chemotherapy. Although they are curative in a low proportion, as a palliative treatment they improve survival rates and the life quality of the patients. In the case of chemotherapy, this has shown limited activity in brain metastasis in the majority of types of tumours. Above all, considering that many chemotherapeutic agents used systematically do not pass through the bbb whereas others can weaken it as they enable the extravasation of the tumour cells in circulation to the brain parenchyma [138].
Breast cancer is the second most common solid neoplasm that metastasises in the brain. Epidemiological studies show that the incidence rates of these metastases are between 10 and 16%, but autopsy reports suggest rates of up to 30% [139, 140]. The HER-2 gene is over-expressed in 20–25% of all breast cancers and makes a well-established risk for brain metastasis [141]. The use of chemotherapy in the treatment of these metastases is not only limited by the lack of drug penetration through the bbb but also by the outward flow of the drugs through the high expression of the P-glycoprotein in the endothelial cells of the brain capillaries [142]. Nevertheless, several agents have shown to have activity in this type of metastasis because of an increase in the permeability of the blood vessels deformed by the metastasis and the effects of radiation [143, 144]. The temozolomide, although it is permeable, has shown a limited activity in brain metastasis [145], but cisplatin has shown clinical activity in this metastasis associated with breast cancer as a single agent and in combination with radiotherapy [146]. Capecitabine is effective in breast cancer as it passes through the bbb barrier with the help of a nucleoside transporter, the hCNT [147]. Other chemotherapeutic agents used in the therapy against brain metastasis in breast cancer, that express activity anti-HER-2, are trastuzumab [148], lapatinib [149], neratinib [150], afatinib [151], pertuzumab [152], and sagopilone [153]. On the other hand, proteins that are associated to hypoxia and angiogenesis have been used as early indicators in the development of patients with metastatic breast cancer that have been previously treated with bevacizumab. In this treatment regimen changes in the levels of IL8 and VEGFR2 can be used to predict the response in these patients [154].
Metastasis in the liver.-In the liver, metastasis are the most frequent malignant processes as almost all the tumours can produce them. The most common ones are those that come from the gastrointestinal tract, especially from the colon and the pancreas, and from the breast and lung are also commonplace. In patients with colon cancer, 40% first suffer from metastasis in the liver and subsequently in the lung. The cells reach the liver through various channels, the portal system, the hepatic artery, the lymphatic vessel of the helium, or by direct extension from nearby organs [155]. Before metastases are detected certain conditions are present that make them possible: the size of the tumour cells, a fenestrated endothelium of the liver, plus the lack of a basal sub-endothelial membrane. The cancer cells extend projections through the fenestrated endothelium in the space of Disse establishing a direct contact with the hepatocytes [156]. The mechanisms that enable the malignant cells in the colon to form hepatic and pulmonary metastases are not well known but clinical evidence shows that different signalling pathways MAPK are involved in this process. The ERK2 activation provides the cells with the capacity to colonise the liver, whilst the reduction of the MAPK p38 signalling gives the cells the capacity to form metastasis in the lung from previously established liver lesions. The reduction in MAPK p38 signalling and the increase in the expression of parathyroid hormone like hormone (PTHLH) contribute with the extravasations of the cells in order to reach the lung. The high activity of PTHLH makes the blood vessels in the lung permeable enough to be passed by the tumour cells in order to colonise the lungs [157, 158]. It has also been shown that the inflammatory response is correlated to the metastatic potential of some tumours in the liver [155].
The liver is the third most common site of breast cancer metastasis and untreated liver metastasis shorten patient survival times to just four to eight months. In breast cancer, TNF-α can trigger e-selectin expression, increasing liver sinusoidal endothelial adhesiveness. This process is similar to the one reported for colorectal and lung cancer [159]. IL-6 induction in breast cancer cell lines reduces cellular adhesion which is associated with reduced e-cadherin expression. This finding correlates with observations of breast carcinoma patients with liver metastasis who express high levels of IL-6. Breast carcinoma cells create a pro-inflammatory microenvironment by secreting cytokines which promote adhesiveness and invasiveness in the liver [160]. Breast carcinoma cells also express chemokine receptors, such as CXCR4, while metastatic cells in the liver express CXCL12, the ligand of CXCR4. This indicates that CXCL12/CXCR4 interaction contributes to liver metastasis. CXCR4 also participates in the modulation of liver metastasis by interacting with integrins [161]. Malignant breast cells, however, also express high levels of CD44 with the highest expression occurring in cells that metastasise to the liver [162]. High levels of claudin-2 expression have also been detected in metastatic breast cells in the liver. These cells metastasise by adhering to extracellular matrix proteins, such as fibronectin and type IV collagen, which are abundant in the liver [163].
In primary colon tumours, aberrant TGFα/EGFR expression helps propagate tumour cells in lymph nodes and the liver [164]. Colon cancer cells express high amounts of EGFR and overexpress TGFα in response to hypoxia, triggering a signalling cascade that helps them survive. Cellular proliferation thus involves Ras/MAPK and anti-apoptotic activity (phosphatidylinositol-3-kinase [PI3K] / Akt) which ultimately correlates with metastasis and resistance to chemotherapy [165, 166]. ER, PR, and HER-2 status is also essential to determining the response to any kind of treatment whether adjuvant hormonal therapy, directed molecular therapy, or chemotherapy. Researchers have evaluated and established the relationship of ER, PR, and HER-2 in primary breast cancer and metastatic cancer in specific organs [167]. For example, one study found that increased HER-2 phosphorylation is a critical factor in establishing liver metastasis in breast cancer [168].
Chemotherapy and hormones remain alternatives for treating breast cancer patients with liver metastasis. Surgery is another option for palliating metastatic complications, and although it is limited to individual liver segments only. Nonetheless, partial removal of the liver offers the possibility of a cure. HER-2-positive invasive ductal carcinoma patients with liver metastasis have achieved complete response through a combination of treatments. These patients took docetaxel and two anti-HER-2 agents, trastuzumab and pertuzumab, along with surgery to resect the metastatic liver part [169]. Another option for treating patients with liver metastasis is to administer a combination of drugs via hepatic arterial infusion (HAI): irinotecan, bevacizumab, and oxaliplatin combined with bevacizumab or cetuximab [170] have been used. In colorectal cancer patients with liver and lung metastases, completely resecting metastases in both organs offers the best survival results. A retrospective study compared general three- and five-year survival rates in three groups of colorectal cancer patients with liver and lung metastases. Patients who received chemotherapy and had surgery to resect only liver metastasis had higher survival rates than patients who only received chemotherapy. The former group, however, had a lower survival rate than patients who received chemotherapy and had surgery to resect both liver and lung metastases [171, 172]. An experimental model demonstrated the involvement of laminin receptor LRP/LR in cancer progression. This model established the role of LRP/LR in adhesion and invasion in HUH-7 liver cancer cells. These cells showed high levels of LRP/LR compared to K562 leukaemia cells and MCF-7 breast cells. Treating liver cells with the anti-LRP/LR specific antibody IgG1-iS18 significantly reduced the adhesive potential of liver cells to laminin-1 and their invasive potential. This result suggests the use the IgG1-iS18 antibody as a therapeutic alternative in liver metastasis [173]. Another study used an animal model to examine the combined effect of SiARN targeting the hGHR growth hormone receptor and 5-fluorouracil on colon cancer metastases. The results of that study showed that this combination reduced the incidence of liver metastasis in colon cancer [174]. A study of 46,027 patients examined metastatic distribution patterns in colon and rectal cancer. This study found that the incidence of liver metastasis was 13.8% in colon cancer patients versus 12.3% in those with rectal cancer. The incidence of lung metastasis was 5.6% in rectal cancer patients and 3.7% in those with colon cancer. The incidence of bone metastasis in rectal cancer patients was 1.2% and 0.8% for colon cancer patients. This study also examined the incidence of bone metastasis in colorectal cancer patients. Incidence was 10.0% in patients with lung metastasis, but 4.5% in patients without lung metastasis. Brain metastasis rates were 3.1% and 0.1%, respectively. Understanding metastatic distribution patterns can help clinicians apply chemotherapy more effectively, and is also valuable for possible surgical treatment [175]. Another study examined the benefits of surgical resection in patients with stage IV metastatic colon cancer. Patients received several cycles of neoadjuvant chemotherapies, such as combinations of folinic acid, fluorouracil (5-FU), and oxaliplatin (FOLFOX); folinic acid, (5-FU), and irinotecan (FOLFIRI); folinic acid, (5-FU), oxaliplatin, and irinotecan (FOLFOXIRI); and bevacizumab, panitumumab, and cetuximab. Patients who were candidates for immediate surgery did not benefit from treatment, while patients who were not candidates for surgery showed a survival benefit from chemotherapy [176]. Another study evaluated the efficacy of treating colon cancer patients with liver metastasis using electromagnetic waves. This treatment was effective in increasing patient survival times and controlling liver metastasis measuring 3 cm or less [177].
Researchers now use models that aim to mimic morphological structures and the relationships that exist in living systems. This requires conducting studies using three-dimensional models that approximate the cellular and molecular composition of tumours. Researchers grow multicellular tumour spheroids, or MTS [178], seed porous scaffolds [179], and create miniature versions of various organs connected by vascular canals called ‘Chips’ [180]. Such models will help researchers develop therapies that act on tissues and systems and not just at the cellular and molecular levels. These advances will make research and clinical practice more efficient, optimise prevention, and create better treatments.
Conclusions
Based on new research findings, the ‘seed and soil’ hypothesis has helped cancer researchers to reassess and create a new framework for analysing metastasis. We now have a more advanced perspective on this complex and multifactorial disease. We see that cancer and its malignancies do not result solely from specific genetic mutations in tumour cells. Moreover, we recognise the importance of studying the tumour macroenvironment in order to achieve a comprehensive understanding of the manifestations of cancer and how to treat it. Any study that focuses only on cells and genes will thus be incomplete as not all people with altered genes develop cancer. Current accepted treatments based on conventional protocols have poor efficacy as they focus on destroying tumours without evaluating cancer patients more comprehensively. Clinicians use the same treatments for all patients with the same histological type of tumour, not considering that tumour progression varies for all patients. We must consider that surrounding tissues consisting of non-cancerous cells including blood vessels and immune system cells play an important role in cancer. Patients require treatments that also focus on the tumour environment in order to develop preventive therapies that make that environment adverse for tumour cells. We must therefore understand the tumour environment in order to clarify our view of this disease and help us use treatments more rationally.
References
1. Varmus H and Weiberg RA (1993) Genes and the Biology of Cancer Scientific American Library, A división of HPHLP, New York
2. Nowell PC (1976) The clonal evolution of tumor cell populations Science 194(4260) 23–28
3. Arvelo F, Cotte C and Sojo F (2014) Células Madre y Cáncer. Invest Clín 55(4) 371–91
4. Sell S and Pierce GB (1994) Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers Lab Invest 70(1) 6–22
5. Arvelo F and Poupon MF (2001) Aspectos Moleculares y Celulares de la Metástasis Cancerosa Acta Cient Venez 52(4) 304–12
6. De Vita VT, Hellman S and Rosemberg SA (2004) Cancer, Principles & Practice of Oncology Lippincott & Wilkins
7. Nguyen DX, Bos PD and Massagué J (2009) Metastasis: from dissemination to organ-specific colonization Nat Rev Cancer 9(4) 274–84
8. Nguyen DX (2001) Tracing the origins of metastasis J Pathol 223(2) 195–204
9. Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation Cell 144(5) 646–74
10. Arvelo F, Sojo F and Cotte C (2016) Tumour progression and metastasis Ecancermedicalscience 10 617 DOI: 10.3332/ecancer.2016.617
11. Ferrarra N, Hans-Peter G and LeCouter J (2003) The biology of VEGF and its receptors Nat Med 9(6) 669–76
12. Guadamillas MC, Cerezo A and del Pozo MA (2011) Overcoming anoikis – pathways to anchorage-independent growth in cancer J Cell Sci 124(Pt 19) 3189–97
13. Carmeliet P (2005) Angiogenesis in life, Disease and Medicine Nature 438 (7070) 932–6
14. Greene HS and Harvey EK (1964) The relationship between the dissemination of tumor cells and the distribution of metastases Cancer Res 24 799–811
15. Auerbach R et al (1987) Specificity of adhesion between murine tumor cells and capillary endothelium: an in vitro correlate of preferential metastasis in vivo Cancer Res 47(6) 1492–6
16. Willmott N and Newton J (1987) Demonstration of site-specific tumor growth by a 50% end point assay Invasion and Metastasis 70(1) 30–40
17. Hiratsuka S et al (2011) Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin upregulation Proc Natl Acad Sci U S A 108(9) 3725–30
18. Kim YJ et al (1999) Distinct selectin ligands on colon carcinoma mucins can mediate pathological interactions among platelets, leukocytes, and endothelium Am J Pathol 155(2) 461–72
19. Padua D et al (2008) TGF beta primes breast tumors for lung metastasis seeding through angiopoietin-like 4 Cell 133(1) 66–77
20. Gupta GP et al (2007) Mediators of vascular remodelling co-opted for sequential steps in lung metastasis Nature 446(7137) 765–770
21. Labelle M, Begum S and Hynes RO (2011) Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis Cancer Cell 20(5) 576–90
22. Gil-Bernabe AM et al (2012) Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice Blood 119(13) 3164–75
23. Qian B et al (2009) A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth PLoS One 4(8) e6562
24. Kaplan RN et al (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche Nature 438(7069) 820–7
25. Deng J et al (2012) S1PR1-STAT3 Signaling is crucial for myeloid cell colonization at future metastatic sites Cancer Cell 21(5) 642–54
26. Hiratsuka S et al (2006) Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis Nat Cell Biol 8(12) 1369–75
27. Ewing J (1928) Neoplastic Diseases 3rd ed WB Saunders; Philadelphia
28. Paget S (1989) The distribution of secondary growths in cancer of the breast Cancer Metastasis Rev 8(2) 98–101
29. Sugarbaker EV, Cohen AM and Ketcham AS (1971) Do metastases metastasize? Ann Surg 174(2) 161–6
30. Hart IR and Fidler IJ (1980) Role of organ selectivity in the determination of metastatic patterns of B16 melanoma Cancer Res 40(7) 2281–7
31. Fidler IJ and Nicolson GL (1976) Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines. J Natl Cancer Inst 57(5) 1199–202
32. Muralidharan-Chari V et al (2010) Microvesicles: mediators of extracellular communication during cáncer progression J Cell Sci 123(Pt 10) 1603–11
33. Martins VR, Dias MS and Hainaut P (2013) Tumor-cell-derived microvesicles as carriers of molecular information in cancer Curr Opinn Oncol 25(1) 66–75
34. Ji H et al (2013) Proteome profiling of exosomes derived from human primary and metastatic colorectal cancer cells reveal differential expression of key metastatic factors and signal transduction components Proteomics13(10–11) 1672–86 DOI: 10.1002/pmic.201200562
35. Zhang HG and Grizzle WE (2014) Exosomes: a novel pathway of local and distant intercellular communication that facilitates the growth and metastasis of neoplastic lesions Am J Pathol 184(1) 28–41 DOI: 10.1016/j.ajpath.2013.09.027
36. Lai RC, Chen TS and Lim SK (2011) Mesenchymal stem cell exosome: a novel stem cell-based therapy for cardiovascular disease Regen Med 6(4) 481–92
37. Skog J et al (2008) Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers Nat Cell Biol 10(12) 1470–6 DOI: 10.1038/ncb1800
38. Thompson CA, Purushothaman A and Ramani VC (2013) Heparanaseregulates secretion,composition and function of tumor cell-derived exosomes J Biol Chem 288(14) 10093–9 DOI: 10.1074/jbc.C112.444562
39. Cho JA et al (2011) Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts Gynecol Oncol 123(2) 379–86 DOI: 10.1016/j.ygyno.2011.08.005
40. Al-Nedawi K, Meehan B and Kerbel RS (2009) Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR Proc Natl Acad Sci USA 106(10) 3794–9 DOI: 10.1073/pnas.0804543106
41. Peinado H et al (2012) Melanoma exosomas educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET Nat Med 18(6) 883–891 DOI: 10.1038/nm.2753
42. Al-Nedawi K et al (2008) Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells Nat Cell Biol 10(5) 619–24 DOI: 10.1038/ncb1725
43. Grange C et al (2011) Microvesicles released from human renal cáncer stem cells stimulate angiogenesis and formation of lung premetastatic niche Cancer Res 71(15) 5346–56 DOI: 10.1158/0008-5472.CAN-11-0241
44. Liu Y et al (2014) Microvesicles secreted from human multiple myeloma cells promote angiogenesis Acta Pharmacol Sin 35(2) 230–8 DOI: 10.1038/aps.2013.141
45. Aga M et al (2014) ExosomalHIF1 alpha supports invasive potential of nasopharyngeal carcinoma associated LMP1-positive exosomes Oncogene 33(37) 4613–22 DOI: 10.1038/onc.2014.66
46. Kobayashi M, Salomon C and Tapia J (2014) Ovarian cancer cell invasiveness is associated with discordant exosomal sequestration of Let- 7 miRNA and miR-200 J Transl Med 12 4 DOI: 10.1186/1479-5876-12-4
47. Soldevilla B et al (2014) Tumor-derived exosomas are enriched in DeltaNp73, which promotes oncogenic potential in acceptor cells and correlates with patient survival Hum Mol Genet 23(2) 467–8 DOI: 10.1093/hmg/ddt437
48. Wang J et al (2014) Bone marrow stromal cell-derived exosomas as communicators in drug resistance in multiple myeloma cells Blood 124(4) 555–66 DOI: 10.1182/blood-2014-03-562439
49. Castellana D et al (2009) Membrane microvesicles as actors in the establishment of a favorable prostatic tumoral niche: a role for activated fibroblasts and CX3CL1-CX3CR1 axis Cancer Res 69(3) 785–93
50. Vella LJ (2014) The emerging role of exosomes in epithelial–mesenchymal-transition in cancer Front Oncol 4361 DOI: 10.3389/fonc.2014.00361
51. Hoshino A (2015) Tumour exosome integrins determine organotropic metastasis Nature 527(7578) 329–35 DOI: 10.1038/nature15756
52. Grange C (2011) Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res 71(15) 5346–56
53. Jung T (2009) CD44v6 dependence of premetastatic niche preparation by exosomes Neoplasia 11(10) 1093–105
54. Costa-Silva B (2015) Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver Nat Cell Biol 17(6) 816–26 DOI: 10.1038/ncb3169
55. Coleman RE (2006) Clinical features of metastatic bone disease and risk of skeletal morbidity Clin Cancer Res 12(20 Pt 2) 6243s–6249s
56. Huang Q and Ouyang X (2012) Biochemical-markers for the diagnosis of bone metastasis: a clinical review Cancer Epidemiol 36(1) 94–8
57. Shiao SL, Chu GC and Chung LW (2016) Regulation of prostate cancer progression by the tumor microenvironment Cancer Lett 380(1)340–8 pii: S0304-3835(15)00773–9 DOI: 10.1016/j.canlet.2015.12.022
58. Psaila B and Lyden D (2009) The metastatic niche: adapting the foreign soil Nat Rev Cancer 9(4) 285–93
59. Kaplan RN, Rafii S and y Lyden D (2006) “Preparing the “soil”: the premetastatic niche” Cancer Res 66(23) 11089–93
60. Sun YX et al (2007). “Expression and activation of alpha v beta 3 integrins by SDF-1/CXC12 increases the aggressiveness of prostate cancer cells” Prostate 67(1) 61–73
61. Roodman GD (2004) Mechanisms of bone metastasis N Engl J Med 350(16) 1655–64
62. Käkönen SM and Mundy GR (2003) Mechanisms of osteolytic bone metastases in breast carcinoma Cancer 97(3 suppl) 834–9
63. Buijs JT and van Der Pluijm G (2009) Osteotropic cancers: from primary tumor to bone Cancer Lett 273(2) 177–93
64. Bendre MS et al (2002) Expression of interleukin 8 and not parathyroid hormone-related protein by human breast cancer cells correlates with bone metastasis in vivo Cancer Res 62(19) 5571–9
65. Cox TR (2015) The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase Nature 522(7554) 106–10
66. Ibrahim T et al (2010) Pathogenesis of osteoblastic bone metastases from prostate cancer Cancer 116(6) 1406–18
67. Theriault RL (2012) Biology of bone metastases Cancer Control 19(2) 92–101
68. Mishra S et al (2011) Blockade of transforming growth factor-beta (TGF-β) signaling inhibits osteoblastic tumorigenesis by a novel human prostate cancer cell line Prostate 71(13) 1441–54
69. Wan X et al (2012) Effect of transforming growth factor beta (TGF-beta) receptor I kinase inhibitor on prostate cancer bone growth Bone 50(3) 695–703
70. Festuccia C, Angelucci A and Gravina GL (2000) Osteoblast-derived TGF-beta1 modulates matrix degrading protease expression and activity in prostate cancer cells Int J Cancer 85(3) 407–15
71. Guise TA et al (2006) Basic mechanisms responsible for osteolytic and osteoblastic bone metastases Clin Cancer Res 12(20 Pt 2) 6213s–6s
72. Bensalah K et al (2008) New circulating biomarkers for prostate cancerProstate Cancer Prostatic Dis 11(2) 112–20
73. Marques RB et al (2010) “Bypass mechanisms of the androgen receptor pathway in therapy-resistant prostate cancer cell models”PLoS One 5(10) e135009
74. Watson PA et al (2010) Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor Proc Natl Acad Sci USA 107(39) 16759–65
75. Body JJ, Casimiro S and Costa L (2015) Targeting bone metastases in prostate cancer: improving clinical outcome Nat Rev Urol Jun;12(6) 340–56 Doi: 10.1038/nrurol.2015.90
76. Zhang G et al (2015) Androgen receptor splice variants circumvent AR blockade by microtubule-targeting agents Oncotarget 6(27) 23358–71 Epub ahead of print
77. Arichi N et al (2015) Versican is a potential therapeutic target in docetaxel-resistant prostate cancer Oncoscience 2(2) 193–204 eCollection 2015
78. Ning YM et al (2010) Phase II trial of bevacizumab, thalidomide, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer J ClinOncol 28(12) 2070–6
79. Qi P et al (2015) A Meta-analysis and indirect comparison of endothelin a receptor antagonist for castration-resistant prostate cancer PLoS One 10(7) e0133803 DOI: 10.1371/journal.pone.0133803
80. Smith MR et al (2015) Denosumab for the prevention of skeletal complications in metastatic castration-resistant prostate cancer: comparison of skeletal-related events and symptomatic skeletal events Ann Oncol 26(2) 368–74
81. Felibert P, Quintana J and Arvelo F (2009) Metaloproteasas. Propiedades, Funciones y sus usos como blancos terapéuticos en el tratamiento antineoplásico Acta Cient Venez 60 11–21
82. Ide H et al (2014) Circulating tumor cell count during zoledronic acid treatment in men with metastatic prostate cancer: a pilot study Prostate Int 2(3) 147–51
83. Hoy SM (2013) Abiraterone acetate: a review of its use in patients with metastatic castration-resistant prostate cancer Drugs 73(18) 2077–91
84. Wen SM, Quan CY and Jiang N (2015) What is the next generation therapeutic strategy for castration-resistant prostate cancer Asian J Androl 17(2) 223–4
85. Li XX et al (2015) Expression of molecular factors correlated with metastasis in small cell lung cancer and their significance Int J Clin Exp Pathol 8(11) 14676–84
86. Nicolson GL (1988) Organ specificity of tumor metastasis. Rote of preferential adhesion, invasion and growth of malignant cells specific secondary sites Cancer Metastasis Rev 7 1432–5
87. Duda DG and Jain RK (2010) Premetastatic lung “niche”: is vascular endothelial growth factor receptor 1 activation required? Cancer Res 70(14) 5670–3
88. Kim S et al (2009) Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis Nature 457(7225) 102–106
89. Hiratsuka S et al (2002) MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis Cancer Cell 2(4) 289–300
90. Dawson MR et al (2009) VEGFR1 activity modulates myeloid cell infiltration in growing lung metastases but is not required for spontaneous metastasis formation PLoS One 4(9) e6525. DOI: 10.1371/journal.pone.0006525
91. Fischer C et al (2008) FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy?Nat Rev Cancer 8(12) 942–56
92. Ferrara N, Gerber HP and LeCouter J (2003) The biology of VEGF and its receptors Nat Med 9(6) 669–76
93. Miao HQ and Klagsbrun M (2000) Neuropilin is a mediator of angiogenesis Cancer Metastasis Rev 19(1–2) 29–37
94. Fischer C et al (2008)FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat Rev Cancer 8(12) 942–56
95. Erler JT et al (2009) Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche Cancer Cell 15(1) 35–44
96. Langley RR et al (2001) Endothelial expression of vascular cell adhesion molecule-1 correlates with metastatic pattern in spontaneous melanoma Microcirculation 8(5) 335–45
97. De Vita VT, Hellman S and Rosemberg SA (2004) Cancer, Principles & Practice of Oncology Lippincott & Wilkins
98. Alexander ET et al (2015) Thrombin inhibition and cyclophosphamide synergistically block tumor progression and metastasis Cancer Biol Ther 16(12) 1802–11 [Epub ahead of print].
99. Bartholomeusz C et al (2015) MEK inhibitor selumetinib (AZD6244; ARRY-142886) prevents lung metastasis in a triple-negative breast cancer xenograft model Mol Cancer Ther 14(12) 2773–81pii: molcanther.0243.2015. [Epub ahead of print]
100. Yoshida T et al (2014) Eribulinmesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states Br J Cancer 110(6) 1497–505 DOI: 10.1038/bjc.2014.80
101. Li JT et al (2015) MiRNA-101 inhibits breast cancer growth and metastasis by targeting CX chemokine receptor 7 Oncotarget 6(31) 30818–30 [Epub ahead of print]
102. Nayak L, Lee EQ and Wen PY (2012) Epidemiology of brain metastases Curr Oncol Rep 14(1) 48–54
103. Brastianos HC, Cahill DP and Brastianos PK (2015) Systemic therapy of brain metastases Curr Neurol Neurosci Rep15(2) 518 DOI: 10.1007/s11910-014-0518-9
104. Fox BD et al (2011) Epidemiology of metastatic brain tumors Neurosurg Clin N Am 22(1) 1–6
105. Sperduto PW et al (2012) Summary report on the graded prognostic assessment: an accurate and facile diagnosis-specific tool to estimate survival for patients with brain metastases J ClinOncol 30(4) 419–25
106. Delattre JY et al (1988) Distribution of brain metastases Arch Neurol 45(7) 741–4
107. Weil RJ et al (2005) Breast cancer metastasis to the central nervous system Am J Pathol 167(4) 913–20
108. Ahmad A et al (2014) Up-regulation of microRNA-10b is associated with the development of breast cancer brain metastasis Am J Transl Res 6(4) 384–90
109. Berghoff AS et al (2014) Differential role of angiogenesis and tumor cell proliferation in brain metastases according to primary tumor type: analysis of 639 cases Neuropathol Appl Neurobiole 41(2) e41–55pub ahead of print 10.1111/nan.12185
110. Pinhel I et al (2012) NCRI Adjuvant breast cancer trial management group: ER and HER2 expression are positively correlated in HER2 non-overexpressing breast cancer Breast Cancer Res 14(2) R46
111. Niikura N et al (2012) Loss of human epidermal growth factor receptor 2 (HER2) expression in metastatic sites of HER2-overexpressing primary breast tumors J ClinOncol 30(6) 593–9
112. Montagna E et al (2009) Central nervous system metastases in a cohort of metastatic breastcancer patients treated with trastuzumab Cancer Chemother Pharmacol 63 275–280
113. Medress Z and Gephart MH (2015) Molecular and genetic pregictors of breast-to-brain metástasis: review and case Presentation Cureus 7(1) e246 DOI 10.7759/cureus.246
114. Gupta P and Srivastava SK (2014) HER2 mediated de novo production of TGFβ leads to SNAIL driven epithelial-to-mesenchymal transition and metastasis of breast cancer Mol Oncol 8(8) 1532–47
115. Kalluri R and Weinberg RA (2009) The basics of epithelial-mesenchymal transition J Clin Invest 119(6) 1420–8
116. Heyn C et al (2006) In vivo MRI of cancer cell fate at the single-cell level in a mouse model of breast cancer metastasis to the brain Magn Reson Med 56(5) 1001–10
117. Carbonell WS et al (2009) The vascular basement membrane as “soil” in brain metastasis PLoS One 4(6):e5857 DOI: 10.1371/journal.pone.0005857
118. Fidler IJ et al (2010) The brain microenvironment and cancer metastasis Mol Cells 30(2) 93–8
119. Lee KY et al (2011) Human brain endothelial cell-derived COX-2 facilitates extravasation of breast cancer cells across the blood-brain barrier Anticancer Res 31(12) 4307–13
120. Malin D et al (2014) αB-crystallin: a novel regulator of breast cancer metastasis to the brainClin Cancer Res20(1) 56–67
121. Zhao C et al (2014) Dysregulation of JAM-A plays an important role in human tumor progression Int J ClinExpPathol 7(10) 7242–8
122. Bos PD et al (2009) Genes that mediate breast cancer metastasis to the brainNature 459(7249) 1005–9
123. Wang L et al (2013) Astrocytes directly influence tumor cell invasion and metastasis in vivo PLoS One4 8(12) e80933 DOI: 10.1371/journal.pone.0080933
124. Sofroniew MV and Vinters HV (2010) Astrocytes: biology and pathologyActa Neuropathol 119(1) 7–35
125. Valiente M et al (2014) Serpins promote cancer cell survival and vascular co-option in brain metastasis Cell 156(5) 1002–16
126. Neman J et al (2014) Human breast cancer metastases to the brain display GABAergic properties in the neural niche Proc Natl Acad Sci USA 111(3) 984–9
127. Winkler F (2014) The brain microenvironment: friend or foe for metastatic tumor cells? Neuro Oncol 16(12) 1565–6
128. Chen J et al (2015) Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain Cancer Res 75(3) 554–65
129. Rojas Laimito K et al (2016) Characterization of the triple negative breast cancer phenotype associated with the development of central nervous system metastases Ecancer 10 632 DOI: 10.3332/ecancer.2016.632
130. Sartorius CA et al (2015) Estrogen promotes the brain metastatic colonization of triple negative breast cancer cells via an astrocyte-mediated paracrine mechanism Oncogene 28 1–12 DOI: 10.1038/onc.2015.353 PMID: 26411365
131. Lockman PR et al (2010) Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer Clin Cancer Res 16(23) 5664–78
132. Gerstner ER and Fine RL (2007) Increased permeability of the blood-brain barrier to chemotherapy in metastatic brain tumors: establishing a treatment paradigm J Clin Oncol 25(16) 2306–12
133. Bronger H et al (2005) ABCC drug efflux pumps and organic anion uptake transporters in human gliomas and the blood-tumor barrier Cancer Res 65(24) 11419–28
134. Bertossi M et al (1997) Ultrastructural and morphometric investigation of human brain capillaries in normal and peritumoral tissues Ultrastruct Pathol 21(1) 41–9
135. Hasegawa H, Ushio Y and Hayakawa T (1983) Changes of the blood-brain barrier in experimental metastatic brain tumors J Neurosurg 59(2) 304–10
136. Liu LB et al (2008) Bradykinin increases blood-tumor barrier permeability by down-regulating the expression levels of ZO-1, occludin, and claudin-5 and rearranging actin cytoskeleton J Neurosci Res 86(5) 1153–68
137. Abbott NJ et al (2010) Structure and function of the blood-brain barrier Neurobiol Dis 37(1) 13–25
138. Blecharz KG et al (2015) Control of the blood-brain barrier function in cancer cell metastasis Biol Cell107(10) 342–71 DOI: 10.1111/boc.201500011 [Epub ahead of print]
139. Patanaphan V, Salazar OM and Risco R (1988) Breast cancer: metastatic patterns and their prognosis South Med J 81(9) 1109–12
140. Lee YT (1983) Breast carcinoma: pattern of metastasis at autopsy J Surg Oncol 23(3) 175–180
141. Slamon DJ et al (1989) Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer Science 244(4905) 707–12
142. Tosoni A, Franceschi E and Brandes AA (2008) Chemotherapy in breast cancer patients with brain metastases: have new chemotherapic agents changed the clinical outcome? Crit Rev Oncol Hematol 68(3) 212–21
143. vanVulpen M et al (2002) Changes in blood-brain barrier permeability induced by radiotherapy: implications for timing of chemotherapy? Oncol Rep 9(4) 683–8
144. Fidler IJ (2011) The role of the organ microenvironment in brain metastasis Semin Cancer Biol 21107–112
145. Siena S et al (2010) Dose-dense temozolomide regimen for the treatment of brain metastases from melanoma, breast cancer, or lung cancer not amenable to surgery or radiosurgery: a multicenter phase II study Ann Oncol 21(3) 655–61
146. Cassier PA et al (2008) A phase 2 trial of whole-brain radiotherapy combined with intravenous chemotherapy in patients with brain metastases from breast cancer Cancer 113(9) 2532–8
147. Chargari C et al (2009) Concurrent capecitabine and whole-brain radiotherapy for treatment of brain metastases in breast cancer patients J Neurooncol 93(3) 379–84
148. Dijkers EC et al (2010) Biodistribution of 89Zr-trastuzumab and PET imaging of HER2-positive lesions in patients with metastatic breast cancer Clin Pharmacol Ther 87(5) 586–92
149. Metro G et al (2011) Clinical outcome of patients with brain metastases from HER2-positive breast cancer treated with lapatinib and capecitabine Ann Oncol 22(3) 625–30
150. Burstein HJ et al (2010) Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer J ClinOncol 28(8) 1301–7
151. Lin NU et al (2012) A phase II study of afatinib (BIBW 2992), an irreversible ErbB family blocker, in patients with HER2-positive metastatic breast cancer progressing after trastuzumab Breast Cancer Res Treat 133(3) 1057–65
152. Moya-Horno I and Cortés J (2015) The expanding role of pertuzumab in the treatment of HER2-positive breast cancer Breast Cancer (Dove Med Press) 7 125–32
153. Freedman RA et al (2011) A phase II study of sagopilone (ZK 219477; ZK-EPO) in patients with breast cancer and brain metastases Clin Breast Cancer 11(6) 376–83
154. Lam SW et al (2016) Angiogenesis- and hypoxia-associated proteins as early indicators of the outcome in patients with metastatic breast cancer given first-line bevacizumab-based therapy Clin Cancer Res 22(7) 1611–20
155. Ma R et al (2015) Mechanisms involved in breast cancer liver metastasis J Transl Med 13 64 DOI: 10.1186/s12967-015-0425-0
156. Martin MD et al (2010) Rapid extravasation and establishment of breast cancer micrometastases in the liver microenvironment Mol Cancer Res 8(10) 1319–27
157. Urosevic J et al (2014) Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signalling and PTHLH Nat Cell Biol 16(7) 685–94
158. del Barco Barrantes I and Nebreda AR (2012) Roles of p38 MAPKs in invasion and metastasis Biochem Soc Trans 40(1) 79–84
159. St Hill CA (2011) Interactions between endothelial selectins and cancer cells regulate metastasis Front Biosci 16 3233–51
160. Asgeirsson KS et al (1998) The effects of IL-6 on cell adhesion and e-cadherin expression in breast cancer Cytokine 10(9) 720–8
161. Furusato B et al (2010) CXCR4 and cancer PatholInt 60(7) 497–505
162. Erin N et al (2013) Differential characteristics of heart, liver, and brain metastatic subsets of murine breast carcinoma Breast Cancer Res Treat 139(3) 677–89
163. Tabaries S et al (2012) Claudin-2 promotes breast cancer liver metastasis by facilitating tumor cell interactions with hepatocytes Mol Cell Biol 32(15) 2979–91
164. Sasaki T, Hiroki K and Yamashita Y (2013) The role of epidermal growth factor receptor in cancer metastasis and microenvironment Biomed Res Int 546318 DOI: 10.1155/2013/546318
165. Herbst RS (2004) Review of epidermal growth factor receptor biology Int J Radiation Oncology Biology Physics 59(2) 21–6
166. Groenen LC, Nice EC and Burgess AW (1994) Structure-function relationships for the EGF/TGF-α family of mitogens Growth Factors 11(4) 235–57
167. Koo JS, Jung W and Jeong J (2010) Metastatic breast cancer shows different immunohistochemical phenotype according to metastatic site Tumori 96(3) 424–32
168. Liu J et al (2012) Comparison of ER/PR and HER2 status in primary and paired liver metastatic sites of breast carcinoma in patients with or without treatment J Cancer Res Clin Oncol 138(5) 837–42
169. Schoellhammer HF et al (2014) Complete pathologic response of HER2-positive breast cancer liver metastasis with dual anti-HER2 antagonism BMC Cancer 14 242 DOI: 10.1186/1471-2407-14-242
170. Said R et al (2015) Dose-finding study of hepatic arterial infusion of irinotecan-based treatment in patients with advanced cancers metastatic to the liver Invest New Drugs 33(4) 911–20 DOI: 10.1007/s10637-015-0251-5
171. Brouquet A et al (2011) Improved survival after resection of liver and lung colorectal metastases compared with liver-only metastases: a study of 112 patients with limited lung metastatic disease J Am Coll Surg 213(1) 62–9
172. Mise Y et al (2014) Simultaneous lung resection via a transdiaphragmatic approach in patients undergoing liver resection for synchronous liver and lung metastases Surgery 156(5) 1197–203
173. Chetty C et al (2014) Anti-LRP/LR specific antibody IgG1-iS18 impedes adhesion and invasion of liver cancer cells PLoS One5 9(5):e96268 DOI: 10.1371/journal.pone.0096268
174. Zhou D et al (2015) Effect of combination therapy of siRNA targeting growth hormone receptor and 5-fluorouracil in hepatic metastasis of colon cancer Oncol Lett 10(6) 3505–9
175. Qiu M et al (2015) Cosgrove DP, Xu R. Pattern of distant metastases in colorectal cancer: a SEER based study Oncotarget 6(36) 38658–666 DOI: 10.18632/oncotarget.6130
176. Al-Hajeili M et al (2016) Neoadjuvant treatment for surgically resectable metastatic colorectal cancer Oncology (Williston Park) 30(1) 10–6pii: 215235
177. Xu C et al (2015) Radiofrequency ablation in treating colorectal cancer patients with liver metastases Asian Pac J Cancer Prev 16(18) 8559–61
178. Privalova AM et al (2015) Microencapsulated multicellular tumor spheroids as a tool to test novel anticancer nanosized drug delivery systems in vitro J Nanosci Nanotechnol 15(7) 4806–14
179. Hidalgo IA et al (2013) Functional electrospun poly (lactic acid) scaffolds for biomedical applications. Experimental conditions, degradation and biocompatibility studyMol Cell Biomech 10 85–105
180. Del Nero P, Song YH and Fischbach C (2013) Microengineered tumor models: insights & opportunities from a physical sciences-oncology perspective Biomed Microdevices 15(4) 583–93 DOI: 10.1007/s10544-013-9763-y