Cancer-associated thrombotic microangiopathy
Govind Babu K and Gita R Bhat
Department of Medical Oncology, Kidwai Memorial Institute of Oncology, Dr MH Marigowda road, Hombegowdanagar, Bangalore-560029, India
Correspondence to: Gita R Bhat. Email: livewire841@gmail.com
Abstract
Cancer-associated thrombotic microangiopathy refers to a group of disorders characterised by microvascular thrombosis, thrombocytopenia, and ischaemic end-organ damage. Haemolytic uraemic syndrome and thrombotic thrombocytopenic purpura are the two major subtypes. It can be a manifestation of the malignancy itself or a complication of its therapy. The addition of several new drugs to the therapeutic armamentarium of cancer has brought to light several novel causative factors of this hitherto uncommon complication. This review covers the aetiology, pathogenesis, clinical manifestations, complications, and the management of cancer-associated thrombotic microangiopathy. Careful review of the patient’s medical records coupled with the correlation of clinical findings and laboratory reports can help clinch the diagnosis and institute appropriate treatment on time.
Keywords: thrombotic microangiopathy, haemolytic uraemic syndrome, thrombotic thrombocytopenic purpura, gemcitabine, mitomycin-C, haematopoietic stem cell transplant, immunotoxins, anti-VEGF, plasmapheresis
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Published: 28/06/2016; Received: 03/05/2016
Introduction
Thrombotic microangiopathies (TMA) are a group of disorders characterised by disseminated occlusive microvascular thrombosis, thrombocytopenia, and ischaemic end-organ damage, most commonly in kidneys and brain. Haemolytic uraemic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP) are the two main subtypes [1].
TTP is described by a pentad of clinical findings including thrombocytopenia, microangiopathic haemolytic anaemia (MAHA), neurologic deficits, such as seizures, hemiplegia and visual disturbances, renal failure, and fever. HUS is defined as a triad of MAHA with varying degrees of thrombocytopenia and renal failure [1, 2]. Fragmentation of erythrocytes as they pass through clogged arterioles gives rise to Coombs-negative haemolytic anaemia with an elevated schistocyte count. Renal insufficiency invariably occurs in HUS but is milder in TTP, where neurological symptoms often predominate [1]. The pathological features include intra-renal or systemic microvascular thrombi with endothelial swelling and microvascular obstruction [3].
von Willebrand factor (vWF) is initially secreted from Wiebel–Palade bodies as multimers tethered to the endothelial cell surface. Here, they provide glycoprotein Ib receptor sites for platelet adhesion and thrombus formation. ADAMTS13, also called von Willebrand factor-cleaving protease (vWF-cp) regulates thrombus formation by cleaving the multimeric vWF [4, 5]. Severe deficiency of ADAMTS13 (<5% of normal) has been proposed as the key pathogenetic factor for idiopathic TTP [6]. Studies have shown that cancer-associated TMA is not associated with the severe deficiency of ADAMTS13 [7].
History of cancer-associated thrombotic microangiopathy
The term ‘microangiopathic haemolytic anaemia’ was coined by Brain et al in 1962. They observed that ‘widespread intra-capillary and intra-arteriolar eosinophilic granular or amorphous thrombi’ could reduce the lumen of the blood vessels to ‘pin-point size’, leading to mechanical red cell destruction by intra-luminal shearing. They described the appearance of fragmented red cells as ‘burr, triangular, and helmet red cells’ [8]. In their series of 25 patients, the various conditions that caused MAHA were TTP, malignant hypertension, acute renal disease, and metastatic carcinomas. In 1979, Antman et al. in the review on MAHA and cancer proposed that the haemolysis and thrombocytopenia were primarily caused by mechanical obstruction of the vascular lumen by tumour cell emboli. They concluded that a variety of systemic malignancies could cause clinicopathological features of TMA without coagulation abnormalities like disseminated intravascular coagulation [9].
Aetiopathogenesis
In patients with cancer, TMA have been reported as follows:
(1) A manifestation of cancer itself:
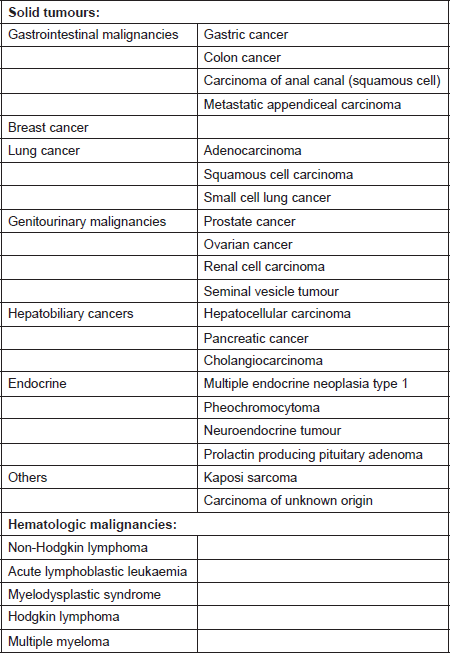
Most cases of cancer-associated TMA have been reported in patients with mucin-producing adenocarcinoma and in those with disseminated malignancies. It has also been described in cases with isolated invasion of the bone marrow by the tumour [10]. The incidence of TMA in this population is said to range from 0.25 to 0.45 persons per million [11]. A prospective study by Lohrmann et al. determined that 5.7% of patients with metastatic carcinoma have MAHA [10]. In an extensive review by Lechner et al., adenocarcinoma was the most common histopathological subtype in patients with cancer-associated TMA. Gastric carcinoma was the most common (26.2%), followed by breast (21.4%), prostate (13.7%), and lung cancer (9.5%) [11]. It has also been reported in pancreatic adenocarcinoma, lymphoma as well as other malignancies [12]. The malignancies associated with MAHA are listed in Table 1 [8, 9, 11, 13–21].
It has been postulated that, along with abnormal angiogenesis in the marrow, aggressive growth of tumours and secondary myelofibrosis may injure the endothelial cell lining of the marrow vasculature by direct invasion. This could result in release of ultra large VWF multimers (ULVWF). In addition, a possible decrease in availability of VWF-cleaving protease (ADAMTS13), possibly through formation of autoantibodies against ADAMTS13 may contribute to the development of TMA. Fragmentation of red blood cells due to direct contact with intraluminal fibrin thrombi or tumour emboli within blood vessels may lead to MAHA [22]. The effect of mucin on endothelial dysfunction has also been proposed as a mechanism for the development of TMA [23].
Table 1. Systemic malignancies associated with microangiopathic haemolytic anaemia [8, 9, 11, 13–21].
(2) As a complication of chemotherapy:
Chemotherapy may cause TMA by two mechanisms, namely an acute immune-mediated reaction or dose-dependent toxicity. Acute and presumed immune-mediated TMA has been reported with oxaliplatin [24]. Oxaliplatin-dependent, platelet-reactive antibodies have been documented in patients with acute oxaliplatin-induced thrombocytopenia [25].
The incidence of gemcitabine induced HUS ranges from 0.015% to 0.31%. The median duration of therapy is 5.8 months, with most of the patients developing HUS within one to two months of the last infusion. It develops after a median cumulative dose of 20 g/m2, with a broad range from 2.5 g/m2 to 48 g/m2. Mortality may be as high as 60% [26–30]. Although gemcitabine-induced TMA is typically dose-dependent, there are case reports of acute TMA following the first or second infusion [31]. It is said to occur due to uncontrolled proximal alternative pathway complement activation. This leads to an increased terminal membrane attack complex giving rise to endothelial cell activation. There is activation of monocytes, neutrophils, and platelet activation and aggregation. Gemcitabine may directly damage endothelial cells, resulting in platelet aggregation and intravascular haemolysis [32].
The incidence of mitomycin-C-induced HUS ranges from 2% to 15%. The risk increases significantly when the cumulative dose exceeds 30 mg/m2 to 40 mg/m2 and after one year of treatment. The prognosis is poor with 75% mortality related to renal failure. Direct endothelial cell injury likely plays a central role. Following endothelial injury and exposure of the subendothelium, activation of platelets and the clotting cascade may occur [27, 28, 30, 33].
The other chemotherapeutic agents that have been implicated in the aetiology of HUS include 5-fluorouracil, bleomycin, cisplatin, cytosine arabinoside, daunomycin, deoxycoformycin, estramustine, and methyl-CCNU [34, 35].
(3) In the setting of bone marrow transplantation:
Chronic kidney disease (CKD) is an important long-term complication of haematopoietic stem cell transplant (HCT), particularly allogeneic in which it develops in 15% to 20% of survivors [36]. Most of the cases of CKD after HCT are thought to be related to a low-grade TMA. However, the incidence in patients undergoing bone marrow transplantation is uncertain, given the difficulty in establishing a diagnosis [37]. It is characterised by slowly rising serum creatinine, hypertension, and disproportionate anaemia. Some cases might have an acute, fulminant presentation with associated renal dysfunction and neurologic manifestations that may represent endothelial damage driven by donor–host interactions [38]. Careful review of previous reports reveals intermittent or persistent elevation of lactate dehydrogenase, low serum haptoglobin, thrombocytopenia, anaemia, and sometimes schistocytosis [39].
Exposure of the kidneys to radiation as a part of the pre-HCT conditioning damages the renal microvasculature. This is the primary cause of renal damage. Concurrent chemotherapy and genetic factors (angiotensin-converting enzyme genotype) play a modulatory role along with post-HCT factors such as use of calcineurin inhibitors (especially sirolimus), the presence of graft versus host disease, the presence of procoagulant state and infection [39–42]. Sirolimus inhibits VEGF resulting in glomerular podocyte injury [43, 44]. Damage to endothelial cells along with cell swelling and fibrin deposition causes microvascular occlusion and results in haemolysis. Glomerular ischaemia gives rise to hypertension and over time leads to fibrosis and renal failure [45].
(4) In those receiving antibodies and immunotoxins:
HUS has been reported in adults and children treated with immunotoxins and is generally reversible [46]. The causative agents include anti-CD 22 immunotoxins CAT-3888, formerly called BL22 [47–49] and moxetumomab pasudotox [50], combotox which is a 2 deglycosylated ricin A chain immunotoxins directed against CD 19 and CD 22 and DAB486IL-2, a recombinant immunotoxin that targets IL-2 [51].
The mechanism of immunotoxin-induced TMA is not well understood. In ricin-induced HUS as demonstrated in animal models, upregulation of pro-inflammatory cytokines MCP-1, TNF-α, IL-1β, and IL-6 promotes an inflammatory environment. This leads to infiltration of the glomeruli with macrophages that stimulate secretion of vWF. This leads to the development of glomerular thrombosis, renal failure, haemolysis, and thrombocytopenia [52, 53]. In case of anti-CD 22 immunotoxins cross reactivity to a different target or non-specific uptake into cells by pinocytosis may play a role [54].
HUS is a dose-limiting toxicity of apolizumab, a humanised monoclonal antibody against an antigen on the β-chain of HLA-DR called 1D10. Alemtuzumab, an anti-CD 52 monoclonal antibody has also been known to induce HUS. It is reversible [55]. Release of cytokines, TNF-α and IL-6 may be causative.
Anti-VEGF therapy
Several anti-VEGF agents, such as bevacizumab [44], sunitinib [56–58], and aflibercept [59], have been associated with TMA, suggesting a potential classwide effect. Eremina et al. have described six biopsy documented cases of TMA-resembling HUS following administration of bevacizumab. These patients developed proteinuria or raised serum creatinine following the initiation of bevacizumab, ultimately leading to renal biopsy. Following discontinuation of therapy, improvement in renal function was noted, suggesting at least partial reversibility.
It has been proposed that inhibition of VEGF in the glomerular microvasculature prevents the formation and maintenance of a healthy, fenestrated endothelium. Without active VEGF signalling, the endothelium is compromised. This leads to loss of integrity of the glomerular filtration barrier [44].
Imatinib
Al Aly et al. have published a case report on a 22-year-old lady with hypereosinophilic syndrome who developed TMA while on Imatinib. ADAMTS13 activity was below the normal range. There was marked elevation of ADAMTS13 inhibitor levels. She was treated with plasma exchange and haemodialysis, leading to haematological recovery but persistent renal impairment [60].
Diagnosing thrombotic microangiopathy
The diagnosis of TMA is based on recognising microvascular thrombosis, red blood cell destruction, and platelet consumption. At present, there are no guidelines to establish the diagnosis.
Blake–Haskins et al. have proposed a detailed grading of TMA. Grade 1 refers to evidence of erythrocyte destruction, without clinical consequences. It has been described as the appearance of schistocytes (≥ 5/HPF) in a patient with no schistocytes at baseline or an increase (≥ 5/HPF) in the frequency of schistocytes over the baseline. Grade 2 includes laboratory findings, such as schistocytosis, drop in haemoglobin by 1 g/dL, and mild renal insufficiency, without clinical consequences. Grade 3 refers to laboratory findings along with clinical consequences. The patient has schistocytosis and drop in haemoglobin by 1 g/dL and either grade 4 thrombocytopenia or moderate renal insufficiency. Grade 4 TMA includes to the presence of life-threatening consequences such as clinically significant haemorrhage requiring intervention, thrombosis, and/or embolism or renal insufficiency requiring intervention. Grade 5 refers to death due to TMA [61].
The clinical features of fulminant HUS/TTP include the classic pentad of MAHA, thrombocytopenia, fever, rapidly progressive renal failure, and neurological deficits [1]. Some may have a subacute presentation with mild thrombocytopenia and gradual deterioration of renal function [1]. Renal dysfunction is seen in almost all the cases and is manifested by elevations in serum creatinine and/or worsening haematuria. Evidence of haemolysis such as fall in haemoglobin, low levels of haptoglobin; elevation of LDH and reticulocyte count and indirect hyperbilirubinaemia is seen frequently. Identification of schistocytes in by peripheral smear may not be documented in all cases.
In the series by Humphreys et al., new or exacerbated hypertension was a prominent feature in gemcitabine-induced HUS. It is important to note that this feature preceded the diagnosis of HUS in all cases. The severity of hypertension correlates with poor outcome [39]. Hypertension in TMA is due to glomerular ischaemia caused by microvascular capillary obstruction [45].
The differences between MAHA caused by systemic malignancy and TTP associated with severe ADAMTS13 deficiency, as per the Oklahoma TTP-HUS registry are as follows [13, 14, 62]:
• Patients with cancer-associated MAHA present at an older ager (mean age: 56 years) versus those with TTP associated with severe ADAMTS13 deficiency (mean age: 40 years).
• We must look for the evidence of active malignancy in those with a history of cancer. Even if the cancer appears to be in remission, evidence of recurrence, such as bone marrow biopsy is necessary.
• Progressive weakness, weight loss, and pain are common in patients with cancer-associated MAHA. They have a longer duration of symptoms (median duration: 21 days) versus those with TTP associated with ADAMTS13 deficiency (median duration: 8 days).
• Pulmonary involvement characterised by dyspnoea, cough, and abnormal chest X-ray is a feature of cancer-associated MAHA.
• Leucoerythroblastic reaction and extreme and extreme elevation of LDH are seen in those with cancer-associated MAHA.
• The median value of ADAMTS13 activity is 50% in those with cancer-associated MAHA.
• MAHA associated with systemic malignancies fails to respond to plasma exchange. On the other hand, patients with TTP due to ADAMTS13 deficiency respond promptly with rapid reversal of neurologic symptoms, reduction of LDH levels over 1 to 2 days and rise in platelet counts over 3 to 4 days.
Is renal biopsy always required?
Renal biopsy is very rarely required, unless the presentation is atypical. The clinical and laboratory features often point to a diagnosis of HUS/TTP. Biopsy findings do not significantly alter management. Biopsy is an invasive procedure and carries increased risk in these patients with thrombocytopenia and other comorbidities. The typical histopathological features are mesangiolysis, basement membrane duplication, glomerular endothelial cell swelling, and tubular injury with interstitial fibrosis [63].
Role of bone marrow biopsy
Patients with history of cancer, including those who appear to be in complete remission need an extensive workup, including bone marrow biopsy to look for the evidence of malignancy [13]. Marrow infiltration by metastatic carcinoma or lymphoma may lead to thrombocytopenia and MAHA [14–16].
Complications
Acute respiratory distress syndrome may develop in patients with a fulminant course. It is associated with a high mortality rate because treatment is even more difficult in a patient with underlying disease [1]. Seizures have been reported in those with the involvement of the central nervous system (CNS). Pancreatic insufficiency may occur, which is manifested as diabetes mellitus, which usually is transient. The muscle, liver, and heart (cardiomyopathy and myocarditis) also can be affected [2]. CKD with hypertension is one of the long-term outcomes [39]. End-stage renal disease (ESRD) may develop.
Differential diagnosis
HUS should be differentiated from isolated renal insufficiency in the presence of myelotoxicity. In the latter scenario, patients usually do not have laboratory evidence of haemolysis with microangiopathy (schistocytes, raised reticulocyte count, indirect hyperbilirubinaemia, elevated lactate dehydrogenase or fibrin split products). Anaemia and thrombocytopenia associated with myelosuppression tend to be more severe. Mild renal insufficiency unrelated to HUS should resolve quickly or return to baseline on rehydration or treatment of the underlying pre-renal state. In renal failure unrelated to HUS, renal biopsy, if performed, will not show the classic microvascular damage with arterioles and small arteries occluded by eosinophilic hyaline thrombi containing fibrin and platelet aggregates [64]. In patients with malignancy, the other causes of thrombocytopenia and haemolysis include disseminated intravascular coagulation and sepsis.
Treatment and outcome
Is there a scope for early detection and prevention of TMA?
In case of gemcitabine induced HUS, early diagnosis may be aided by weekly visits to the chemotherapy day care by the patient might give a chance to detect new or exacerbated hypertension as it develops [65]. Renal shielding during total body irradiation in the setting of allogeneic HCT is said to be protective [66].
As of today, there is no definitive approach for cancer-associated TMA. Treatments comprise of the following main categories:
(1) When a given therapy is implicated as causative, it should be discontinued. Reintroducing the drug at a lower dose may be a strategy to avoid recurrent TMA, while allowing for continued treatment. Retreatment has been successfully carried out with CAT-3888, DAB486IL-2, and bevacizumab. However, it may be difficult to choose an appropriate dose for reintroduction and the risks and benefits involved need to be carefully assessed [44, 49, 67].
(2) Immunocomplex removal: This includes plasmapheresis, immunoadsorption, haemodialysis, or exchange transfusion. Although the efficacies of plasma exchange and immunoadsorption chromatography are arguable, either or both should be initiated promptly at diagnosis in all patients. These may need to be continued for months [11].
(3) Immunosuppressive therapies: The value of corticosteroids is uncertain [1].
(4) Miscellaneous: Blood pressure should be controlled.
(5) Eculizumab: It is a recombinant humanised monoclonal antibody that binds to complement C5 protein, inhibiting its cleavage and thus preventing the generation of the terminal complement attack complex C5b-9.The common adverse events are headache, anaemia, and diarrhoea. Neisseria meningitides vaccination is indicated at least two weeks prior to treatment. It was initially approved for the treatment of paroxysmal nocturnal haemoglobinuria and has also been used in atypical HUS.
Al Ustwani et al. have reported the use of eculizumab in four patients with gemcitabine-induced HUS. In their series, patients were initiated upon eculizumab as the patients did not show improvement of renal function or haemolysis even after administration of corticosteroids stopping gemcitabine for three to five weeks. It resulted in the resolution of microangiopathic haemolysis and thrombocytopenia in all four patients. All patients had significant improvement in renal function but did not return to baseline. None of the patients had serious adverse events [32].
Conclusion
The diagnosis of cancer-associated TMA needs a high index of suspicion. Careful review of diagnosis of cancer, treatment history and interpretation of subtle clinical signs will aid in clinching the diagnosis. Patients with smaller tumour burden, or those in whom anticancer therapy is effective, and in cases where vigilance has led to early diagnosis tend to do better. It has also been suggested that prognosis is better when TMA occurs as a manifestation of the underlying cancer than as a complication of therapy.
Abbreviations used in this article
ADAMTS13 – A disintegrin-like and metalloprotease with thrombospondin type-1 repeats
CKD – Chronic kidney disease
HUS – Haemolytic uraemic syndrome
MAHA – Microangiopathic haemolytic anaemia
TMA – Thrombotic microangiopathy
TTP – Thrombotic thrombocytopenic purpura
ULVWF multimers – Ultra large von Willebrand factor multimers
vWF – von Willebrand factor
vWF-cp – von Willebrand factor-cleaving protease
References
1. Fojo AT Metabolic emergencies In: Devita, Hellman and Rosenberg’s cancer: Principles and practice of oncology, edited by DeVita, Vincent T, Hellman S, Lawrence TS and Rosenberg SA, Wolters Kluwer, 10th edition, pp 1822–1831
2. Corrigan JJ and Boineau FG (2001) Hemolytic-uremic syndrome Pediatr Rev 22 365–369 PMID: 11691946
3. Moake JL (2002) Thrombotic microangiopathies N Eng J Med 347 589–600 DOI: 10.1056/NEJMra020528
4. Ruggeri ZM (2007) The role of von Willebrand factor in thrombus formation Thromb Res 120[Suppl 1] S5–S9 DOI: 10.1016/j.thromres.2007.03.011 PMID: 17493665 PMCID: 2702526
5. Sadler JE (1998) Biochemistry and genetics of von Willebrand factor Annu Rev Biochem 67 395–424 DOI: 10.1146/annurev.biochem.67.1.395 PMID: 9759493
6. Bianchi V, Robles R and Alberio L et al (2002) Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: a severely deficient activity is specific for thrombotic thrombocytopenic purpura Blood 100 710–713 DOI: 10.1182/blood-2002-02-0344 PMID: 12091372
7. Fontana S, Gerritsen HE and Hovinga JK et al (2001) Microangiopathic haemolytic anaemia in metastasizing malignant tumours is not associated with a severe deficiency of the von Willebrand factor-cleaving protease Br J Haematol 113 100–102 DOI: 10.1046/j.1365-2141.2001.02704.x PMID: 11328288
8. Brain MC, Dacie JV and Hourihane OB (1962) Microangiopathic hemolytic anemia: the possible role of vascular lesions in pathogenesis Br J Haematol 8 358–374 DOI: 10.1111/j.1365-2141.1962.tb06541.x PMID: 14014893
9. Antman KH, Skarin AT and Mayer RJ et al (1979) Microangiopathic hemolytic anemia and cancer: a review Medicine 58 377–384 DOI: 10.1097/00005792-197909000-00004 PMID: 481196
10. Lohrmann HP, Adam W and Heymer B et al (1973) Microangiopathic hemolytic anemia in metastatic carcinoma: report of eight cases Ann Intern Med 79 368–375 DOI: 10.7326/0003-4819-79-3-368 PMID: 4748253
11. Lechner K and Obermeier HL (2012) Cancer-related microangiopathic hemolytic anemia. Clinical and laboratory features in 168 reported cases Medicine 91 1–11 DOI: 10.1097/MD.0b013e3182603598
12. Mungall S and Mathieson P (2002) Hemolytic uremic syndrome in metastatic adenocarcinoma of the prostate Am J Kidney Dis 40 1334–1336 DOI: 10.1053/ajkd.2002.36929 PMID: 12460056
13. George JN (2011) Systemic malignancies as a cause of unexpected microangiopathic hemolytic anemia and thrombocytopenia Oncology 25(10) 908–914 PMID: 22010388
14. Francis KK, Kalyanam N and Terrell DR et al (2007) Disseminated malignancy misdiagnosed as thrombotic thrombocytopenic purpura: a report of 10 cases and a systematic review of the literature The Oncologist 12 11–19 DOI: 10.1634/theoncologist.12-1-11
15. Oberic L, Buffet M and Schwarzinger M et al (2009) Cancer awareness in atypical thrombotic microangiopathies The Oncologist 14 769–779 DOI: 10.1634/theoncologist.2009-0067 PMID: 19684072
16. Elliott MA, Letendre L and Gastineau DA et al (2010) Cancer-associated microangiopathic hemolytic anemia with thrombocytopenia: an important diagnostic consideration Eur J Haematol 85 43–50 PMID: 20331741
17. Neidhardt B, Hartwich G and Preiss G et al (1979) EMicroangiopathic hemolytic anemia in metastasizing appendiceal carcinoma Med Welt 30 1822Y1826
18. Kouides PA, Phatak PD and Cramer SF (1996) Fatal thrombotic thrombocytopenic purpura (TTP) presenting concurrently with metastatic multiple endocrine neoplasia (MEN) type I Hematopathol Mol Hematol 10 161Y170
19. Kovacs K and Garvey MB (2003) Thrombotic thrombocytopenic purpura associated with a prolactin-producing pituitary adenoma Am J Hematol 74 55Y59 DOI: 10.1002/ajh.10366
20. Sakai C, Takagi T and Oguro M et al (1994) Malignant pheochromocytoma accompanied by microangiopathic hemolytic anemia: a case report Jpn J Clin Oncol 24 171Y174
21. Schweizer H, Boehm J and Winterer JT et al (2010) Phaeochromocytoma and thrombotic microangiopathy: favourable outcome despite advanced renal failure J Clin Pathol 63 754Y756 DOI: 10.1136/jcp.2010.077511
22. Brain MC, Azzopardi JG and Baker LR et al (1970) Microangiopathic haemolytic anaemia and mucin forming adenocarcinoma Br J Haematol 18 183–192 DOI: 10.1111/j.1365-2141.1970.tb01433.x PMID: 5439525
23. Werner TL, Agarwal N and Carney HM et al (2007) Management of cancer-associated thrombotic microangiopathy: what is the right approach? Am J Hematol 82 295–298 DOI: 10.1002/ajh.20783
24. Dahabreh I, Tsoutsos G and Tseligas D et al (2006) Hemolytic uremic syndrome following the infusion of oxaliplatin: case report BMC Clin Pharmacol 6 5–8 DOI: 10.1186/1472-6904-6-5 PMID: 16968538 PMCID: 1574347
25. Curtis BR, Kaliszewski J and Marques MB et al (2006) Immune-mediated thrombocytopenia resulting from sensitivity to oxaliplatin Am J Hematol 81 193–198 DOI: 10.1002/ajh.20516 PMID: 16493620
26. Flombaum CD, Mouradian JA and Casper ES et al (1999) Thrombotic microangiopathy as a complication of long-term therapy with gemcitabine Am J Kidney Dis 33 555–562 DOI: 10.1016/S0272-6386(99)70194-0 PMID: 10070921
27. Pisoni R, Ruggenenti P and Remuzzi G (2001) Drug-induced thrombotic microangiopathy: incidence, prevention and management Drug Saf 24 491–501 DOI: 10.2165/00002018-200124070-00002 PMID: 11444722
28. Dlott JS, Danielson CF and Blue-Hnidy DE et al (2004) Drug-induced thrombotic thrombocytopenic purpura/hemolytic uremic syndrome: a concise review Ther Apher Dial 8 102–111 DOI: 10.1111/j.1526-0968.2003.00127.x PMID: 15255125
29. Izzedine H, Isnard-Bagnis C and Launay-Vacher V et al (2006) Gemcitabine-induced thrombotic microangiopathy: a systematic review Nephrol Dial Transplant 21 3038–3045 DOI: 10.1093/ndt/gfl507 PMID: 16968717
30. Lesesne JB, Rothschild N and Erickson B et al (1989) Cancer-associated hemolytic-uremic syndrome: analysis of 85 cases from a national registry J Clin Oncol 7 781–789 PMID: 2497229
31. De Smet D, Jochmans K and Neyns B (2008) Development of thrombotic thrombocytopenic purpura after a single dose of gemcitabine Ann Hematol 87 495–496 DOI: 10.1007/s00277-007-0429-9
32. Al Ustwani O, Lohr J and Dy G et al (2014) Eculizumab therapy for gemcitabine induced hemolytic uremic syndrome: case series and concise review J Gastrointest Oncol 1 E30–E33
33. Valavaara R and Nordman E (1985) Renal complications of mitomycin C therapy with special reference to the total dose Cancer 55 47–50 PMID: 3917353
34. Sheldon R and Slaughter D (1986) A syndrome of microangiopathic hemolytic anemia, renal impairment, and pulmonary edema in chemotherapy treated patients with adenocarcinoma Cancer 58 1428–1436 PMID: 3091244
35. Walter RB, Joerger M and Pestalozzi BC (2002) Gemcitabine-associated hemolytic-uremic syndrome Am J Kidney Dis 40 E16 DOI: 10.1053/ajkd.2002.35758 PMID: 12324937
36. Zager RA (1994) Acute renal failure in the setting of bone marrow transplantation Kidney Int 46 1443–1458 DOI: 10.1038/ki.1994.417 PMID: 7853806
37. Verburgh CA, Vermeij CG and Zijlmans JM et al (1996) Haemolytic uraemic syndrome following bone marrow transplantation. Case report and review of the literature Nephrol Dial Transplant 11 1332–1337 DOI: 10.1093/ndt/11.7.1332 PMID: 8672033
38. Martinez MT, Bucher Ch and Stussi G et al (2005) Transplant-associated microangiopathy (TAM) in recipients of allogeneic hematopoietic stem cell transplants Bone Marrow Transplant 36 993–1000 DOI: 10.1038/sj.bmt.1705160 PMID: 16184183
39. Humphreys BD, Soiffer RJ and Magee CC (2005) Renal failure associated with cancer and its treatment: an update J Am Soc Nephrol 16 151–161 DOI: 10.1681/ASN.2004100843
40. Miralbell R, Bieri S and Mermillod B et al (1996) Renal toxicity after allogeneic bone marrow transplantation: the combined effects of total-body irradiation and graft-versus-host disease J Clin Oncol 14 579–585 PMID: 8636774
41. Lawton CA, Fish BL and Moulder JE (1994) Effect of nephrotoxic drugs on the development of radiation nephropathy after bone marrow transplantation Int J Radiat Oncol Biol Phys 28 883–889 DOI: 10.1016/0360-3016(94)90108-2 PMID: 8138441
42. Henry NL, Li S and Kim HT et al (2004) Sirolimus and thrombotic microangiopathy after allogeneic stem cell transplantation J Am Soc Nephrol 15 578A
43. Cutler C, Henry N and Magee C et al (2005) Sirolimus and thrombotic microangiopathy after allogeneic hematopoietic stem cell transplantation Biol Blood Marrow Transplant 11 551–557 DOI: 10.1016/j.bbmt.2005.04.007 PMID: 15983555
44. Eremina V, Jefferson JA and Kowaleska J et al (2008) VEGF inhibiton and renal thrombotic microangiopathy N Eng J Med 358 1129–1136 DOI: 10.1056/NEJMoa0707330
45. Remuzzi G, Ruggenenti P and Bertani T (1994) Thrombotic microangiopathy In: Renal pathology, edited by Tisher CC, Philadelphia, Lippincott, pp 1154–1184
46. Wayne AS, Fitzgerald DJ and Kreitman RJ et al (2014) Immunotoxins for leukemia Blood 123 2470–2477 DOI: 10.1182/blood-2014-01-492256 PMID: 24578503 PMCID: 3990911
47. Kreitman RJ, Squires DR and Stetler-Stevenson M et al (2005) Phase I trial of recombinant immunotoxin RFB4 (dsFv)-PE38 (BL22) in patients with B-cell malignancies J Clin Oncol 23 6719–6729 DOI: 10.1200/JCO.2005.11.437 PMID: 16061911
48. Kreitman RJ, Wilson WH and Bergeron K et al (2001) Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia N Engl J Med 345 241–247 DOI: 10.1056/NEJM200107263450402 PMID: 11474661
49. Kreitman RJ, Stetler-Stevenson M and Margulies I et al (2009) Phase II trial of recombinant immunotoxin RFB4 (dsFv)-PE38 (BL22) in patients with hairy cell leukemia J Clin Oncol 27 2983–2990 DOI: 10.1200/JCO.2008.20.2630 PMID: 19414673 PMCID: 2702232
50. Alderson RF, Kreitman RJ and Chen T et al (2009) CAT-8015: a second-generation pseudomonas exotoxin A-based immunotherapy targeting CD22-expressing hematologic malignancies Clin Cancer Res 15 832–839 DOI: 10.1158/1078-0432.CCR-08-1456 PMID: 19188153 PMCID: 2742326
51. Messmann RA, Vitetta ES and Headlee D et al (2000) A phase I study of combination therapy with immunotoxins IgG-HD37-deglycosylated ricin A chain (dgA) and IgG-RFB4-dgA (Combotox) in patients with refractory CD19(þ), CD22(þ) B cell lymphoma Clin Cancer Res 6 1302–1313 PMID: 10778955
52. Taylor CM, Williams JM and Lote CJ et al (1999) A laboratory model of toxin-induced hemolytic uremic syndrome Kidney Int 55 1367–1374 DOI: 10.1046/j.1523-1755.1999.00387.x PMID: 10201001
53. Korcheva V, Wong J and Corless C et al (2005) Administration of ricin induces a severe inflammatory response via nonredundant stimulation of ERK, JNK, and P38 MAPK and provides a mouse model of hemolytic uremic syndrome Am J Pathol 166 323–339 DOI: 10.1016/S0002-9440(10)62256-0 PMID: 15632024 PMCID: 1602309
54. Pastan I, Hassan R and FitzGerald DJ et al (2007) Immunotoxin treatment of cancer Annu Rev Med 58 221–237 DOI: 10.1146/annurev.med.58.070605.115320
55. Lin TS, Stock W and Xu H et al (2009) A phase I/II dose escalation study of apolizumab (Hu1D10) using a stepped-up dosing schedule in patients with chronic lymphocytic leukemia and acute leukemia Leuk Lymphoma 50 1958–1963 DOI: 10.3109/10428190903186486 PMID: 19860603
56. Frangie C, Lefaucheur C and Medioni J et al (2007) Renal thrombotic microangiopathy caused by anti-VEGF-antibody treatment for metastatic renal-cell carcinoma Lancet Oncol 8 177–178 DOI: 10.1016/S1470-2045(07)70037-2 PMID: 17267332
57. Kapiteijn E, Brand A and Kroep J et al (2007) Sunitinib induced hypertension, thrombotic microangiopathy and reversible posterior leukencephalopathy syndrome Ann Oncol 18 1745–1747 DOI: 10.1093/annonc/mdm454 PMID: 17890216
58. Bollee G, Patey N and Cazajous G et al (2009) Thrombotic microangiopathy secondary to VEGF pathway inhibition by sunitinib Nephrol Dial Transplant 24 682–685 DOI: 10.1093/ndt/gfn657
59. Izzedine H, Brocheriou I and Deray G et al (2007) Thrombotic microangiopathy and anti-VEGF agents Nephrol Dial Transplant 22 1481–1482 DOI: 10.1093/ndt/gfl565
60. Al Aly Z, Philoctete Ashley JM and Gellens ME et al (2005) Thrombotic thrombocytopenic purpura in a patient treated with imatinib mesylate: true association or mere coincidence? Am J Kidney Dis 45 762–768 DOI: 10.1053/j.ajkd.2004.12.017 PMID: 15806480
61. Blake-Haskins JA, Lechleider RJ and Kreitman RJ (2011) Thrombotic microangiopathy with targeted cancer agents Clin Cancer Res 17 5858–5866 DOI: 10.1158/1078-0432.CCR-11-0804 PMID: 21813634 PMCID: 3176977
62. George JN (2010) How I treat patients with thrombotic thrombocytopenic purpura—2010 Blood 116 4060–4069 DOI: 10.1182/blood-2010-07-271445 PMID: 20686117
63. Cohen EP, Hussain S and Moulder JE (2003) Successful treatment of radiation nephropathy with angiotensin II blockade Int J Radiat Oncol Biol Phys 55 190–193 DOI: 10.1016/S0360-3016(02)03793-8
64. Fung MC, Storniolo AM and Nguyen B et al (1999) A review of hemolytic uremic syndrome in patients treated with gemcitabine therapy Cancer 85 2023–2032 DOI: 10.1002/(SICI)1097-0142(19990501)85:9<2023::AID-CNCR21>3.0.CO;2-2 PMID: 10223245
65. Humphreys BD, Sharman JP and Henderson JM et al (2004) Gemcitabine associated thrombotic microangiopathy Cancer 100 2664–2670 DOI: 10.1002/cncr.20290 PMID: 15197810
66. Lawton CA, Barber-Derus SW and Murray KJ et al (1992) Influence of renal shielding on the incidence of late renal dysfunction associated with T-lymphocyte deplete bone marrow transplantation in adult patients Int J Radiat Oncol Biol Phys 23 681–686 DOI: 10.1016/0360-3016(92)90035-G PMID: 1612975
67. Foss FM, Borkowski TA and Gilliom M et al (1994) Chimeric fusion protein toxin DAB486IL-2 in advanced mycosis fungoides and the Sezary syndrome: correlation of activity and interleukin-2 receptor expression in a phase II study Blood 84 1765–1774 PMID: 8080984